Chapter 4 Quality Evaluation
Quality control of DADA2 results will help us have more rational determinations on the further data analysis.
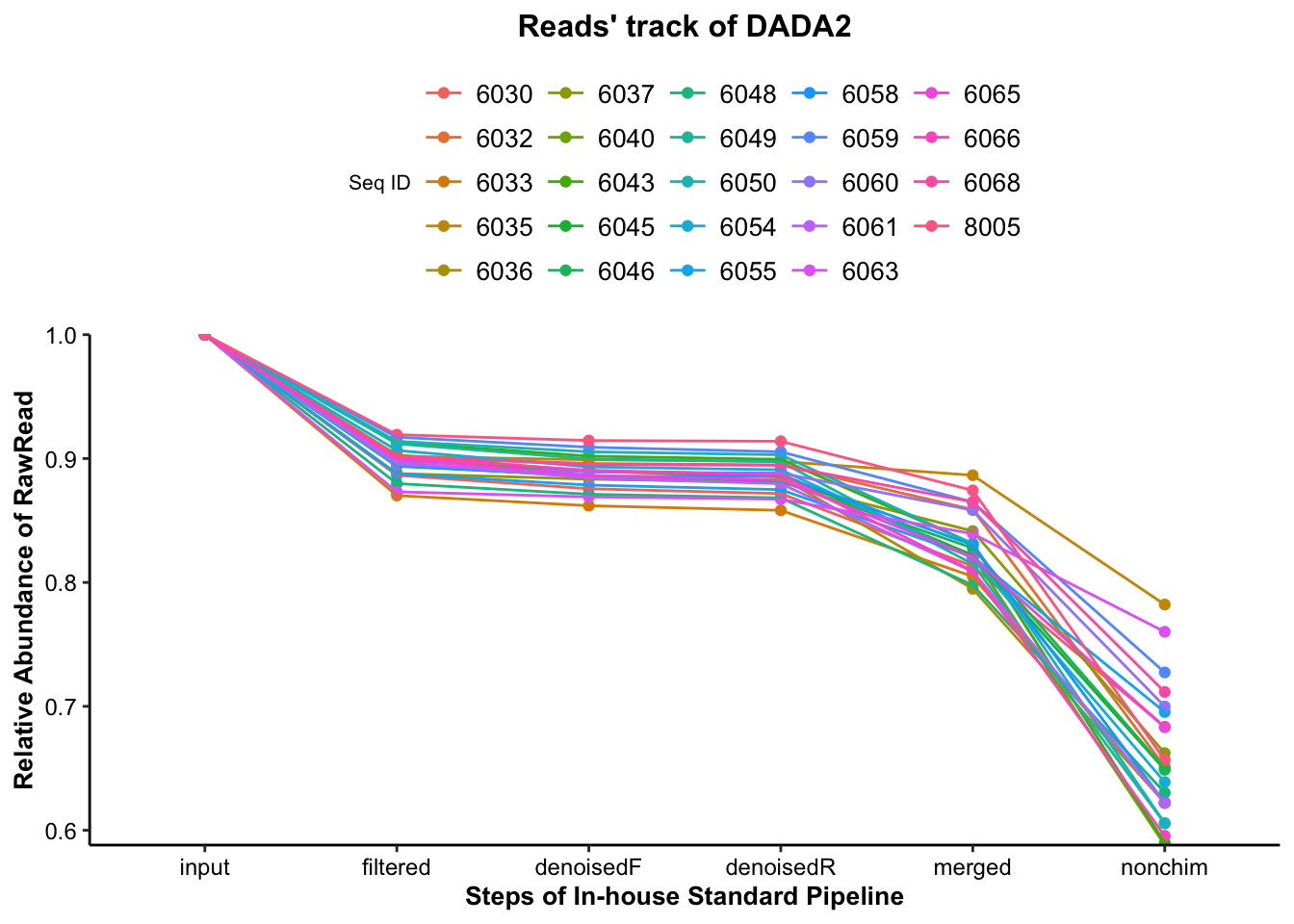
Firstly, the reads’ track of DADA2 could show us the Changed Ratio of reads through the in-house standard amplicon sequencing data upstream pipeline.
Then, the Evaluation of the spike-in samples from the Reference Matrix will reflect the quality of sequencing data.
Thirdly, we suggest you that utilizing the rarefaction curves to assess the sequence depth per sample and choose the rational cutoff of OTU Number to do rarefy.
Finally, obtaining a whole picture on the final phyloseq object.
Outline of this Chapter:
4.3 Reads track
4.3.2 Plotting Metaphlan
This procedure only perform in metagenomic data
How to obtain the input data (use
?plot_metaphlanTrackto get the details)/share/projects/Engineering/pipeline_output/PipelineJob_141157_20220823: the pipeline outputBJ_RoundG_ReadsQC: prefix of resultsresult: output of directory
python /share/projects/Analytics/IO/zouhua/Script/obtain_metaphlan_ReadsQC.py \
-f /share/projects/Engineering/pipeline_output/PipelineJob_141157_20220823 \
-p BJ_RoundG_ReadsQC \
-o result- the results of Reads QC
## SeqID Raw_read1 Raw_read2 fastp_trim1 fastp_trim2 trimmomatic_trim1 trimmomatic_trim2 kneaddata_rmhost1 kneaddata_rmhost2
## 1: 18067 45077982 45077982 44862250 44862250 41821866 41821866 41097049 41097049
## 2: 17849 41421167 41421167 41295206 41295206 40053357 40053357 39210455 39210455
## 3: 18022 42814142 42814142 42610037 42610037 40187608 40187608 39552557 39552557
## 4: 17962 42950934 42950934 42485249 42485249 40274876 40274876 39692768 39692768
## 5: 17912 44754250 44754250 44551046 44551046 41557190 41557190 40830893 40830893
## 6: 17909 42698262 42698262 42491213 42491213 39526119 39526119 38695718 38695718- plotting
## [1] "This palatte have 19 colors!"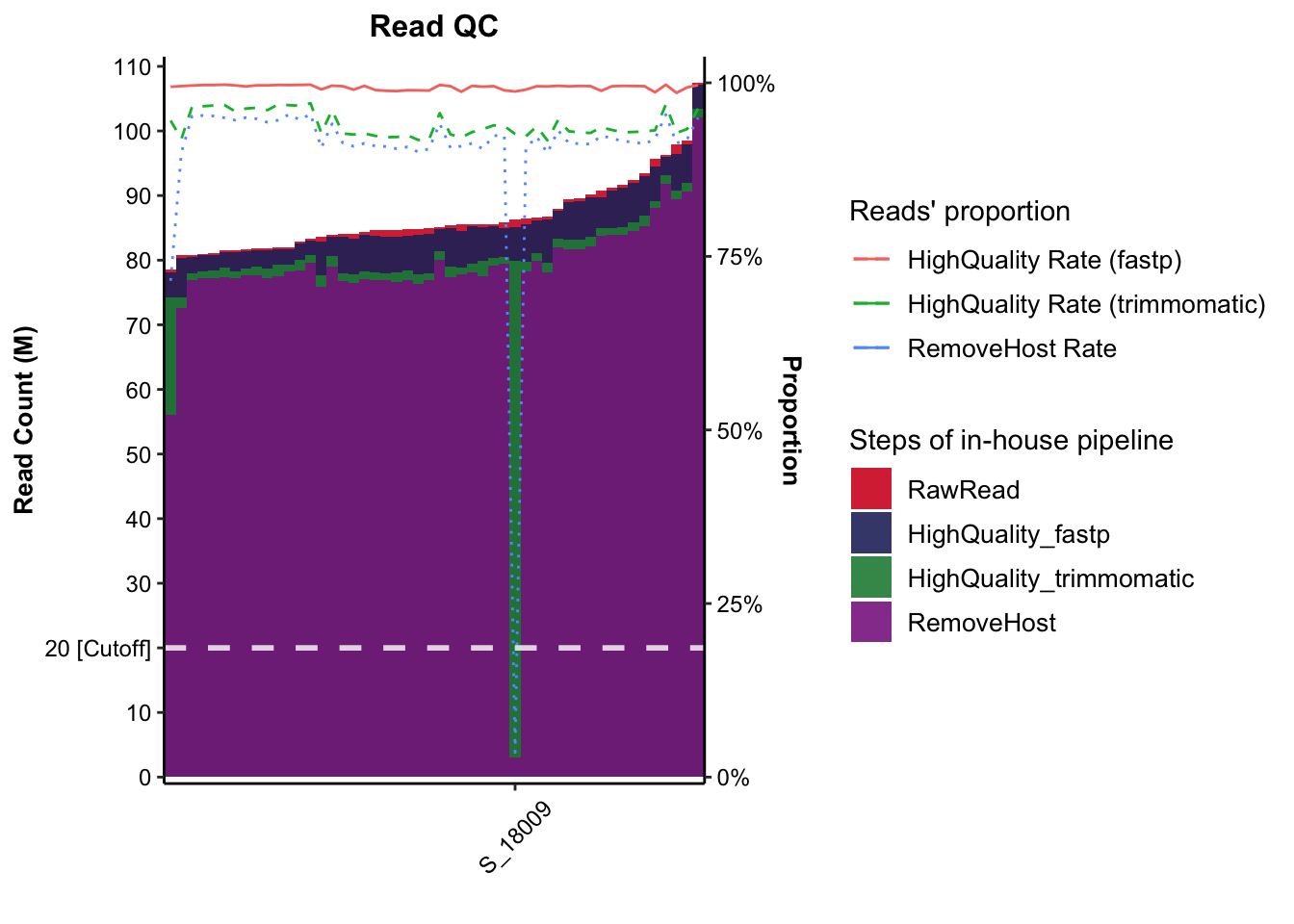
Figure 4.2: Reads’ track of Metaphlan2
4.4 Spike-in (BRS) sample assessment
The spike-in sample is use to evaluate the consistent quality on bacteria when we have multiple sequence batches data. We devised an evaluation system containing the Correlation Coefficient, Bray Curtis Distance and Impurity Level to assess the sequencing data quality.
Please use the default Reference and Save directory to obtain and save the spike-in sample matrix when you run run_RefCheck.
4.4.1 16s data
The taxonomic levels of spike-in sample’s bacteria is genus. Firstly, using the summarize_taxa to get the genus level phyloseq object and get the BRS_ID.
## Group
## S6030 BB
## S6032 BB
## S6033 BB
## S6035 AA
## S6036 BB
## S6037 AA
## S6040 BB
## S6043 AA
## S6045 BB
## S6046 BB
## S6048 BB
## S6049 AA
## S6050 BB
## S6054 BB
## S6055 BB
## S6058 BB
## S6059 AA
## S6060 AA
## S6061 AA
## S6063 BB
## S6065 AA
## S6066 AA
## S6068 BB
## S8005 QCdo run_RefCheck2 under the optimal parameters (Depcrecated run_RefCheck).
BRS_ID: the ID of BRS sample;
Reference: the directory of the latest spike-in sample matrix (default: /share/projects/Analytics/analytics/XMAS/RefCheck/);
Save: the directory to save the latest spike-in sample matrix (default: /share/projects/Analytics/analytics/XMAS/RefCheck/).
To see more details to use ?run_RefCheck2.
dada2_ps_genus_brs_res <-
run_RefCheck2(
ps = dada2_ps_genus,
BRS_ID = "S8005",
Reference = NULL,
Ref_type = "16s",
Save = NULL)
head(dada2_ps_genus_brs_res)## Noting: the Reference Matrix is for 16s
## S8005 is in the Reference Matrix's samples and remove it to run
##
## ############Matched baterica of the BRS sample#############
## The number of BRS' bacteria matched the Reference Matrix is [15]
## g__Bifidobacterium
## g__Bacteroides
## g__Faecalibacterium
## g__Lactobacillus
## g__Parabacteroides
## g__Collinsella
## g__Coprococcus_3
## g__Dorea
## g__Streptococcus
## g__Roseburia
## g__Anaerostipes
## g__Escherichia_Shigella
## g__Enterococcus
## g__Prevotella_9
## g__Eggerthella
##
## The number of the additional bacteria compared to Reference Matrix is [1]
## ###########################################################
##
## ##################Status of the BRS sample##################
## Whether the BRS has the all bateria of Reference Matrix: TRUE
## Correlation Coefficient of the BRS is: 0.9714
## Bray Curtis of the BRS is: 0.07607
## Impurity of Max additional genus (g__Cutibacterium) of the BRS is: 0.06409
## ###########################################################
## #####Final Evaluation Results of the BRS #######
## The BRS of sequencing dataset passed the cutoff of the Reference Matrix
## Cutoff of Coefficient is 0.8946
## Cutoff of BrayCurtis is 0.1425
## Cutoff of Impurity is 0.1565
## ###########################################################
## Gold_Cutoff BRS
## Coef 0.8946 0.97140
## Bray 0.1425 0.07607
## Impurity(max) 1.0000 0.06409We could see that the messages are comprised of four parts.
the 1st part showed the type of reference matrix and whether the spike-in sample had been added to reference matrix;
the 2nd part revealed that what and how many the matched bacterica of the spike-in sample are;
the 3nd part showed that the value of evaluation system in the spike-in sample;
the 4nd part showed that whether the spike-in sample passes the cutoff of evaluation system.
4.4.2 Metagenomic data
The taxonomic levels of spike-in sample’s bacteria is species. Firstly, using the summarize_taxa to get the species level phyloseq object and then do run_RefCheck under the optimal parameters.
Quality Control by spike-in sample in metagenomic
- get the BRS_ID
metaphlan2_ps_species <- summarize_taxa(ps = metaphlan2_ps,
taxa_level = "Species")
metaphlan2_ps_species@sam_data## Group phynotype
## s1 BB 0.00
## s2 AA 2.50
## s3 BB 0.00
## s4 AA 1.25
## s5 AA 30.00
## s6 AA 15.00
## s7 BB 8.75
## s8 BB 0.00
## s9 BB 3.75
## s10 BB 2.50
## s11 BB 15.00
## s12 BB 2.50
## s13 BB 2.50
## s14 BB 0.00
## s15 BB 1.07
## s16 BB 2.50
## s17 AA 5.00
## s18 BB 35.00
## s19 BB 7.50
## s20 BB 15.00
## s21 AA 3.75
## s22 AA 3.75
## refE QC NA- run
run_RefCheck2
metaphlan2_ps_species_brs_res <-
run_RefCheck2(
ps = metaphlan2_ps_species,
BRS_ID = "refE",
Reference = NULL,
Ref_type = "MGS",
Save = NULL)
head(metaphlan2_ps_species_brs_res)## Noting: the Reference Matrix is for MGS
##
## ############Matched baterica of the BRS sample#############
## The number of BRS' bacteria matched the Reference Matrix is [15]
## s__Bifidobacterium_adolescentis
## s__Bifidobacterium_longum
## s__Bifidobacterium_pseudocatenulatum
## s__Collinsella_aerofaciens
## s__Bacteroides_ovatus
## s__Bacteroides_thetaiotaomicron
## s__Bacteroides_uniformis
## s__Bacteroides_vulgatus
## s__Bacteroides_xylanisolvens
## s__Prevotella_copri
## s__Streptococcus_salivarius
## s__Coprococcus_comes
## s__Dorea_formicigenerans
## s__Roseburia_hominis
## s__Faecalibacterium_prausnitzii
## The number of bacteria unmatched the Reference Matrix is [11]
## s__Propionibacterium_acnes
## s__Bifidobacterium_bifidum
## s__Eggerthella_unclassified
## s__Bacteroides_fragilis
## s__Bacteroides_intestinalis
## s__Parabacteroides_goldsteinii
## s__Enterococcus_faecalis
## s__Enterococcus_faecium
## s__Lactobacillus_pentosus
## s__Lactobacillus_salivarius
## s__Escherichia_coli
## The number of the additional bacteria compared to the Reference Matrix is [57]
## ###########################################################
##
## ##################Status of the BRS sample##################
## Whether the BRS has the all bateria of Reference Matrix: FALSE
## Correlation Coefficient of the BRS is: -0.1179
## Bray Curtis of the BRS is: 0.8761
## Impurity of the BRS is: 32.76
## ###########################################################
## #####Final Evaluation Results of the BRS #######
## The BRS of sequencing dataset didn't pass the cutoff of the Reference Matrix
## ###########################################################
## Gold_Cutoff BRS
## Coef 0.8726 -0.1179
## Bray 0.2064 0.8761
## Impurity 6.4400 32.76004.5 Removing BRS
After evaluating the sequencing quality, we remove the BRS.
dada2_ps_remove_BRS <- get_GroupPhyloseq(
ps = dada2_ps,
group = "Group",
group_names = "QC")
dada2_ps_remove_BRS## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 896 taxa and 23 samples ]
## sample_data() Sample Data: [ 23 samples by 1 sample variables ]
## tax_table() Taxonomy Table: [ 896 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 896 tips and 893 internal nodes ]
## refseq() DNAStringSet: [ 896 reference sequences ]4.6 Rarefaction Curves
Rarefaction curves are often used when calculating alpha diversity indices, because increasing numbers of sequenced taxa allow increasingly accurate estimates of total population diversity. Rarefaction curves can therefore be used to estimate the full sample richness, as compared to the observed sample richness.
plot_RarefCurve(ps = dada2_ps_remove_BRS,
taxa_level = "OTU",
step = 400,
label = "Group",
color = "Group")## rarefying sample S6030
## rarefying sample S6032
## rarefying sample S6033
## rarefying sample S6035
## rarefying sample S6036
## rarefying sample S6037
## rarefying sample S6040
## rarefying sample S6043
## rarefying sample S6045
## rarefying sample S6046
## rarefying sample S6048
## rarefying sample S6049
## rarefying sample S6050
## rarefying sample S6054
## rarefying sample S6055
## rarefying sample S6058
## rarefying sample S6059
## rarefying sample S6060
## rarefying sample S6061
## rarefying sample S6063
## rarefying sample S6065
## rarefying sample S6066
## rarefying sample S6068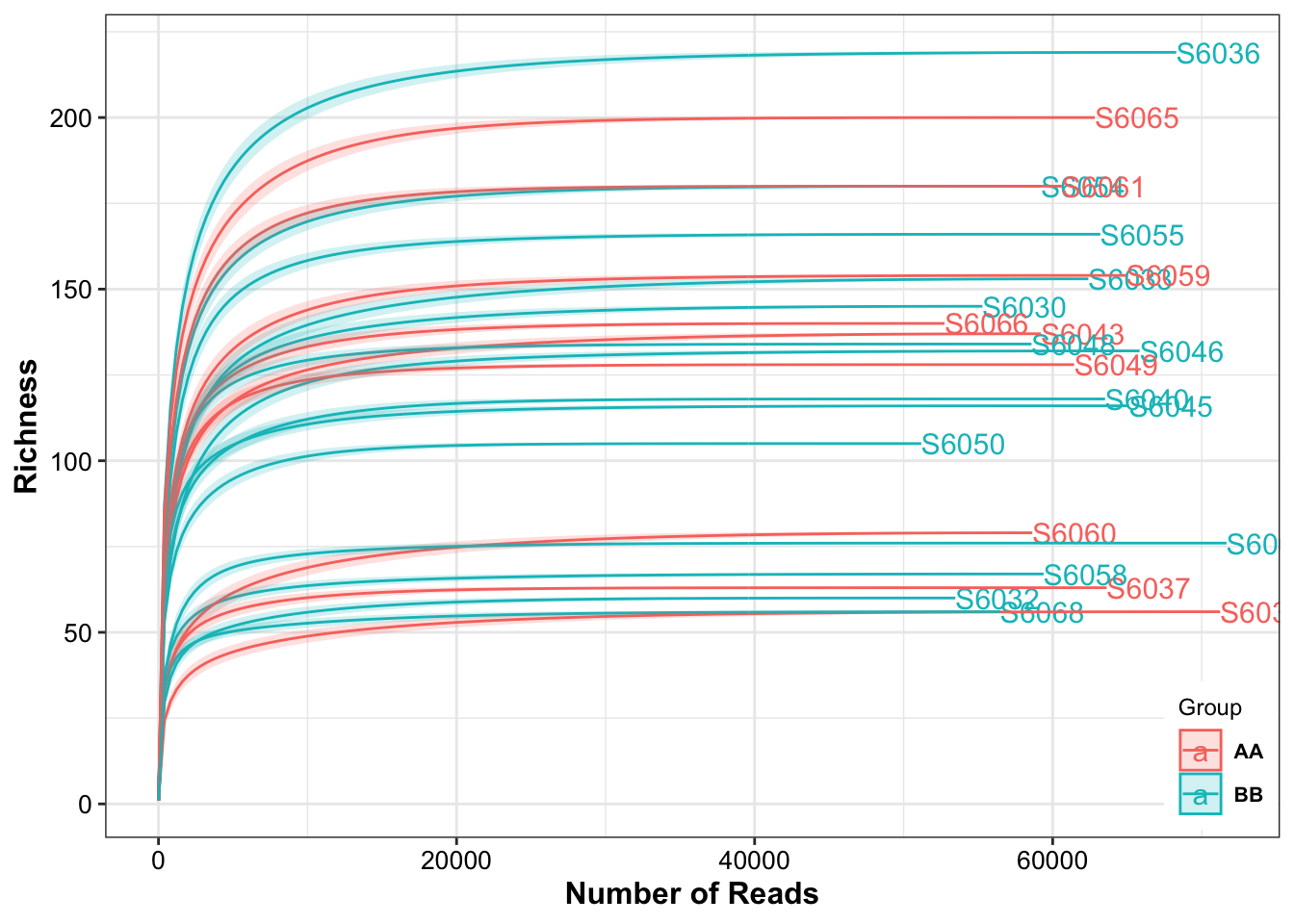
Figure 4.3: Rarefaction Curves
The result showed that all the samples had different sequencing depth but had the full sample richness.
4.7 Summarize phyloseq-class object
Summarizing the phyloseq-class object by using summarize_phyloseq. It displayed that briefly introduction of the object.
summarize_phyloseq(ps = dada2_ps_remove_BRS)
## Compositional = NO2
## 1] Min. number of reads = 511812] Max. number of reads = 716673] Total number of reads = 14089154] Average number of reads = 61257.17391304355] Median number of reads = 614357] Sparsity = 0.8610248447204976] Any OTU sum to 1 or less? YES8] Number of singletons = 49] Percent of OTUs that are singletons
## (i.e. exactly one read detected across all samples)010] Number of sample variables are: 1Group2
## [[1]]
## [1] "1] Min. number of reads = 51181"
##
## [[2]]
## [1] "2] Max. number of reads = 71667"
##
## [[3]]
## [1] "3] Total number of reads = 1408915"
##
## [[4]]
## [1] "4] Average number of reads = 61257.1739130435"
##
## [[5]]
## [1] "5] Median number of reads = 61435"
##
## [[6]]
## [1] "7] Sparsity = 0.861024844720497"
##
## [[7]]
## [1] "6] Any OTU sum to 1 or less? YES"
##
## [[8]]
## [1] "8] Number of singletons = 4"
##
## [[9]]
## [1] "9] Percent of OTUs that are singletons\n (i.e. exactly one read detected across all samples)0"
##
## [[10]]
## [1] "10] Number of sample variables are: 1"
##
## [[11]]
## [1] "Group"The minus account of the OTU counts is 51181 in the phyloseq object, and we can use it as the threshold to rarefy.
Notice the Sparsity (0.865), indicating the data has many zeros and pay attention to the downstream data analysis. A common property of amplicon based microbiota data generated by sequencing.
4.8 Variability
We use the variability to measure measure heterogeneity in OTU/ASV abundance data.
\[Variability_{X} = \frac{sd(X)}{mean(X)}\]
4.8.1 Coefficient of variation
Coefficient of variation (C.V), i.e. sd(x)/mean(x) is a widely used approach to measure heterogeneity in OTU/ASV abundance data. The plot below shows CV-mean(relaive mean abundance) relationship in the scatter plot, where variation is calculated for each OTU/ASV across samples versus mean relative abundance. Now plot C.V.
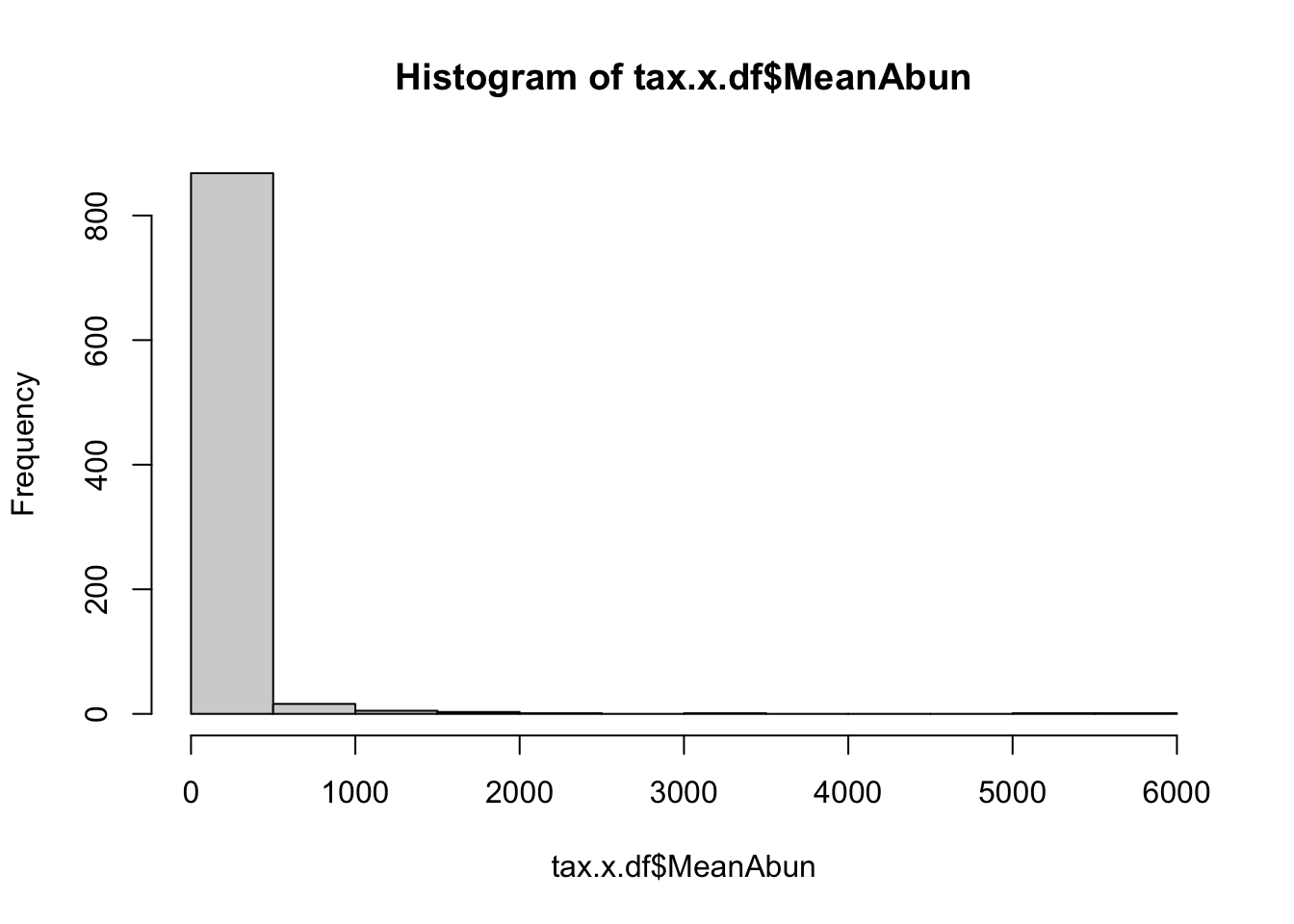
Figure 4.4: Density of the Taxa Mean abundance
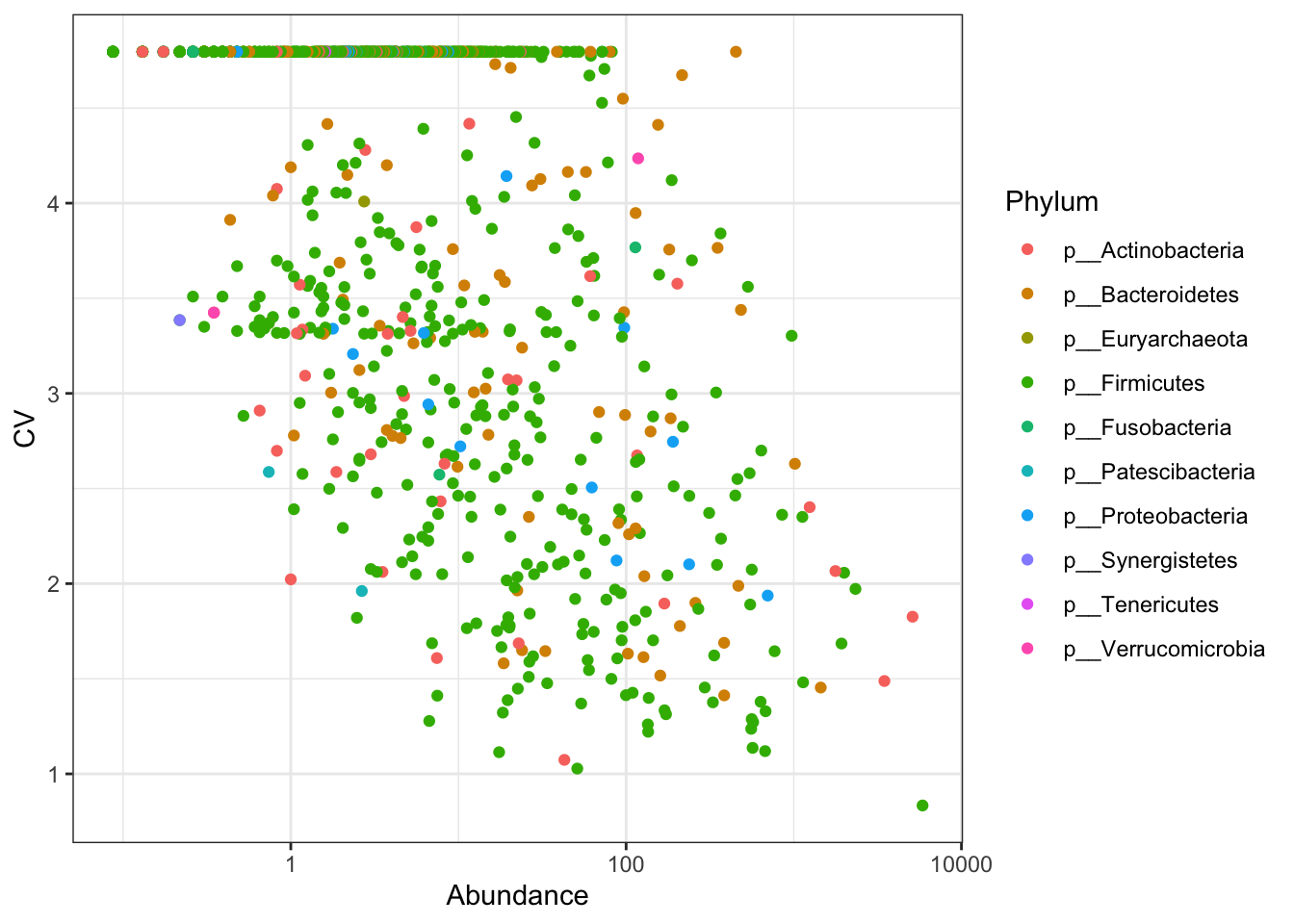
Figure 4.5: Coefficient of variation
From the above two plots, we see that there are several OTUs which have high C.V. and low mean.
4.9 Systematic Information
## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.1.3 (2022-03-10)
## os macOS Monterey 12.2.1
## system x86_64, darwin17.0
## ui RStudio
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz Asia/Shanghai
## date 2023-11-30
## rstudio 2023.09.0+463 Desert Sunflower (desktop)
## pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [2] CRAN (R 4.1.0)
## ade4 1.7-22 2023-02-06 [2] CRAN (R 4.1.2)
## annotate 1.72.0 2021-10-26 [2] Bioconductor
## AnnotationDbi 1.60.2 2023-03-10 [2] Bioconductor
## ape 5.7-1 2023-03-13 [2] CRAN (R 4.1.2)
## backports 1.4.1 2021-12-13 [2] CRAN (R 4.1.0)
## base64enc 0.1-3 2015-07-28 [2] CRAN (R 4.1.0)
## Biobase 2.54.0 2021-10-26 [2] Bioconductor
## BiocGenerics 0.40.0 2021-10-26 [2] Bioconductor
## BiocParallel 1.28.3 2021-12-09 [2] Bioconductor
## biomformat 1.22.0 2021-10-26 [2] Bioconductor
## Biostrings 2.62.0 2021-10-26 [2] Bioconductor
## bit 4.0.5 2022-11-15 [2] CRAN (R 4.1.2)
## bit64 4.0.5 2020-08-30 [2] CRAN (R 4.1.0)
## bitops 1.0-7 2021-04-24 [2] CRAN (R 4.1.0)
## blob 1.2.4 2023-03-17 [2] CRAN (R 4.1.2)
## bookdown 0.34 2023-05-09 [2] CRAN (R 4.1.2)
## broom 1.0.5 2023-06-09 [2] CRAN (R 4.1.3)
## bslib 0.6.0 2023-11-21 [1] CRAN (R 4.1.3)
## cachem 1.0.8 2023-05-01 [2] CRAN (R 4.1.2)
## callr 3.7.3 2022-11-02 [2] CRAN (R 4.1.2)
## car 3.1-2 2023-03-30 [2] CRAN (R 4.1.2)
## carData 3.0-5 2022-01-06 [2] CRAN (R 4.1.2)
## caTools 1.18.2 2021-03-28 [2] CRAN (R 4.1.0)
## checkmate 2.2.0 2023-04-27 [2] CRAN (R 4.1.2)
## cli 3.6.1 2023-03-23 [2] CRAN (R 4.1.2)
## cluster 2.1.4 2022-08-22 [2] CRAN (R 4.1.2)
## coda 0.19-4 2020-09-30 [2] CRAN (R 4.1.0)
## codetools 0.2-19 2023-02-01 [2] CRAN (R 4.1.2)
## coin 1.4-2 2021-10-08 [2] CRAN (R 4.1.0)
## colorspace 2.1-0 2023-01-23 [2] CRAN (R 4.1.2)
## conflicted 1.2.0 2023-02-01 [2] CRAN (R 4.1.2)
## cowplot 1.1.1 2020-12-30 [2] CRAN (R 4.1.0)
## crayon 1.5.2 2022-09-29 [2] CRAN (R 4.1.2)
## data.table 1.14.8 2023-02-17 [2] CRAN (R 4.1.2)
## DBI 1.1.3 2022-06-18 [2] CRAN (R 4.1.2)
## DelayedArray 0.20.0 2021-10-26 [2] Bioconductor
## DESeq2 1.34.0 2021-10-26 [2] Bioconductor
## devtools 2.4.5 2022-10-11 [2] CRAN (R 4.1.2)
## digest 0.6.33 2023-07-07 [1] CRAN (R 4.1.3)
## dplyr * 1.1.2 2023-04-20 [2] CRAN (R 4.1.2)
## DT 0.28 2023-05-18 [2] CRAN (R 4.1.3)
## edgeR 3.36.0 2021-10-26 [2] Bioconductor
## ellipsis 0.3.2 2021-04-29 [2] CRAN (R 4.1.0)
## emmeans 1.8.7 2023-06-23 [1] CRAN (R 4.1.3)
## estimability 1.4.1 2022-08-05 [2] CRAN (R 4.1.2)
## evaluate 0.21 2023-05-05 [2] CRAN (R 4.1.2)
## FactoMineR 2.8 2023-03-27 [2] CRAN (R 4.1.2)
## fansi 1.0.4 2023-01-22 [2] CRAN (R 4.1.2)
## farver 2.1.1 2022-07-06 [2] CRAN (R 4.1.2)
## fastmap 1.1.1 2023-02-24 [2] CRAN (R 4.1.2)
## flashClust 1.01-2 2012-08-21 [2] CRAN (R 4.1.0)
## foreach 1.5.2 2022-02-02 [2] CRAN (R 4.1.2)
## foreign 0.8-84 2022-12-06 [2] CRAN (R 4.1.2)
## Formula 1.2-5 2023-02-24 [2] CRAN (R 4.1.2)
## fs 1.6.2 2023-04-25 [2] CRAN (R 4.1.2)
## genefilter 1.76.0 2021-10-26 [2] Bioconductor
## geneplotter 1.72.0 2021-10-26 [2] Bioconductor
## generics 0.1.3 2022-07-05 [2] CRAN (R 4.1.2)
## GenomeInfoDb 1.30.1 2022-01-30 [2] Bioconductor
## GenomeInfoDbData 1.2.7 2022-03-09 [2] Bioconductor
## GenomicRanges 1.46.1 2021-11-18 [2] Bioconductor
## ggplot2 * 3.4.2 2023-04-03 [2] CRAN (R 4.1.2)
## ggpubr * 0.6.0 2023-02-10 [2] CRAN (R 4.1.2)
## ggrepel 0.9.3 2023-02-03 [2] CRAN (R 4.1.2)
## ggsignif 0.6.4 2022-10-13 [2] CRAN (R 4.1.2)
## glmnet 4.1-7 2023-03-23 [2] CRAN (R 4.1.2)
## glue 1.6.2 2022-02-24 [2] CRAN (R 4.1.2)
## gplots 3.1.3 2022-04-25 [2] CRAN (R 4.1.2)
## gridExtra 2.3 2017-09-09 [2] CRAN (R 4.1.0)
## gtable 0.3.3 2023-03-21 [2] CRAN (R 4.1.2)
## gtools 3.9.4 2022-11-27 [2] CRAN (R 4.1.2)
## highr 0.10 2022-12-22 [2] CRAN (R 4.1.2)
## Hmisc 5.1-0 2023-05-08 [2] CRAN (R 4.1.2)
## htmlTable 2.4.1 2022-07-07 [2] CRAN (R 4.1.2)
## htmltools 0.5.7 2023-11-03 [1] CRAN (R 4.1.3)
## htmlwidgets 1.6.2 2023-03-17 [2] CRAN (R 4.1.2)
## httpuv 1.6.11 2023-05-11 [2] CRAN (R 4.1.3)
## httr 1.4.6 2023-05-08 [2] CRAN (R 4.1.2)
## igraph 1.5.0 2023-06-16 [1] CRAN (R 4.1.3)
## IRanges 2.28.0 2021-10-26 [2] Bioconductor
## iterators 1.0.14 2022-02-05 [2] CRAN (R 4.1.2)
## jquerylib 0.1.4 2021-04-26 [2] CRAN (R 4.1.0)
## jsonlite 1.8.7 2023-06-29 [2] CRAN (R 4.1.3)
## kableExtra 1.3.4 2021-02-20 [2] CRAN (R 4.1.2)
## KEGGREST 1.34.0 2021-10-26 [2] Bioconductor
## KernSmooth 2.23-22 2023-07-10 [2] CRAN (R 4.1.3)
## knitr 1.43 2023-05-25 [2] CRAN (R 4.1.3)
## labeling 0.4.2 2020-10-20 [2] CRAN (R 4.1.0)
## later 1.3.1 2023-05-02 [2] CRAN (R 4.1.2)
## lattice 0.21-8 2023-04-05 [2] CRAN (R 4.1.2)
## leaps 3.1 2020-01-16 [2] CRAN (R 4.1.0)
## libcoin 1.0-9 2021-09-27 [2] CRAN (R 4.1.0)
## lifecycle 1.0.3 2022-10-07 [2] CRAN (R 4.1.2)
## limma 3.50.3 2022-04-07 [2] Bioconductor
## locfit 1.5-9.8 2023-06-11 [2] CRAN (R 4.1.3)
## magrittr 2.0.3 2022-03-30 [2] CRAN (R 4.1.2)
## MASS 7.3-60 2023-05-04 [2] CRAN (R 4.1.2)
## Matrix 1.6-0 2023-07-08 [2] CRAN (R 4.1.3)
## MatrixGenerics 1.6.0 2021-10-26 [2] Bioconductor
## matrixStats 1.0.0 2023-06-02 [2] CRAN (R 4.1.3)
## memoise 2.0.1 2021-11-26 [2] CRAN (R 4.1.0)
## metagenomeSeq 1.36.0 2021-10-26 [2] Bioconductor
## mgcv 1.8-42 2023-03-02 [2] CRAN (R 4.1.2)
## mime 0.12 2021-09-28 [2] CRAN (R 4.1.0)
## miniUI 0.1.1.1 2018-05-18 [2] CRAN (R 4.1.0)
## modeltools 0.2-23 2020-03-05 [2] CRAN (R 4.1.0)
## multcomp 1.4-25 2023-06-20 [2] CRAN (R 4.1.3)
## multcompView 0.1-9 2023-04-09 [2] CRAN (R 4.1.2)
## multtest 2.50.0 2021-10-26 [2] Bioconductor
## munsell 0.5.0 2018-06-12 [2] CRAN (R 4.1.0)
## mvtnorm 1.2-2 2023-06-08 [2] CRAN (R 4.1.3)
## nlme 3.1-162 2023-01-31 [2] CRAN (R 4.1.2)
## nnet 7.3-19 2023-05-03 [2] CRAN (R 4.1.2)
## permute 0.9-7 2022-01-27 [2] CRAN (R 4.1.2)
## phyloseq * 1.38.0 2021-10-26 [2] Bioconductor
## pillar 1.9.0 2023-03-22 [2] CRAN (R 4.1.2)
## pkgbuild 1.4.2 2023-06-26 [2] CRAN (R 4.1.3)
## pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 4.1.0)
## pkgload 1.3.2.1 2023-07-08 [2] CRAN (R 4.1.3)
## plyr 1.8.8 2022-11-11 [2] CRAN (R 4.1.2)
## png 0.1-8 2022-11-29 [2] CRAN (R 4.1.2)
## prettyunits 1.1.1 2020-01-24 [2] CRAN (R 4.1.0)
## processx 3.8.2 2023-06-30 [2] CRAN (R 4.1.3)
## profvis 0.3.8 2023-05-02 [2] CRAN (R 4.1.2)
## promises 1.2.0.1 2021-02-11 [2] CRAN (R 4.1.0)
## ps 1.7.5 2023-04-18 [2] CRAN (R 4.1.2)
## purrr 1.0.1 2023-01-10 [2] CRAN (R 4.1.2)
## R6 2.5.1 2021-08-19 [2] CRAN (R 4.1.0)
## RColorBrewer 1.1-3 2022-04-03 [2] CRAN (R 4.1.2)
## Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.1.3)
## RCurl 1.98-1.12 2023-03-27 [2] CRAN (R 4.1.2)
## remotes 2.4.2 2021-11-30 [2] CRAN (R 4.1.0)
## reshape2 1.4.4 2020-04-09 [2] CRAN (R 4.1.0)
## rhdf5 2.38.1 2022-03-10 [2] Bioconductor
## rhdf5filters 1.6.0 2021-10-26 [2] Bioconductor
## Rhdf5lib 1.16.0 2021-10-26 [2] Bioconductor
## rlang 1.1.1 2023-04-28 [1] CRAN (R 4.1.2)
## rmarkdown 2.23 2023-07-01 [2] CRAN (R 4.1.3)
## rpart 4.1.19 2022-10-21 [2] CRAN (R 4.1.2)
## RSQLite 2.3.1 2023-04-03 [2] CRAN (R 4.1.2)
## rstatix 0.7.2 2023-02-01 [2] CRAN (R 4.1.2)
## rstudioapi 0.15.0 2023-07-07 [2] CRAN (R 4.1.3)
## rvest 1.0.3 2022-08-19 [2] CRAN (R 4.1.2)
## S4Vectors 0.32.4 2022-03-29 [2] Bioconductor
## sandwich 3.0-2 2022-06-15 [2] CRAN (R 4.1.2)
## sass 0.4.6 2023-05-03 [2] CRAN (R 4.1.2)
## scales 1.2.1 2022-08-20 [2] CRAN (R 4.1.2)
## scatterplot3d 0.3-44 2023-05-05 [2] CRAN (R 4.1.2)
## sessioninfo 1.2.2 2021-12-06 [2] CRAN (R 4.1.0)
## shape 1.4.6 2021-05-19 [2] CRAN (R 4.1.0)
## shiny 1.7.4.1 2023-07-06 [2] CRAN (R 4.1.3)
## stringi 1.7.12 2023-01-11 [2] CRAN (R 4.1.2)
## stringr 1.5.0 2022-12-02 [2] CRAN (R 4.1.2)
## SummarizedExperiment 1.24.0 2021-10-26 [2] Bioconductor
## survival 3.5-5 2023-03-12 [2] CRAN (R 4.1.2)
## svglite 2.1.1 2023-01-10 [2] CRAN (R 4.1.2)
## systemfonts 1.0.4 2022-02-11 [2] CRAN (R 4.1.2)
## TH.data 1.1-2 2023-04-17 [2] CRAN (R 4.1.2)
## tibble * 3.2.1 2023-03-20 [2] CRAN (R 4.1.2)
## tidyr 1.3.0 2023-01-24 [2] CRAN (R 4.1.2)
## tidyselect 1.2.0 2022-10-10 [2] CRAN (R 4.1.2)
## urlchecker 1.0.1 2021-11-30 [2] CRAN (R 4.1.0)
## usethis 2.2.2 2023-07-06 [2] CRAN (R 4.1.3)
## utf8 1.2.3 2023-01-31 [2] CRAN (R 4.1.2)
## vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.1.3)
## vegan 2.6-4 2022-10-11 [2] CRAN (R 4.1.2)
## viridisLite 0.4.2 2023-05-02 [2] CRAN (R 4.1.2)
## webshot 0.5.5 2023-06-26 [2] CRAN (R 4.1.3)
## withr 2.5.0 2022-03-03 [2] CRAN (R 4.1.2)
## Wrench 1.12.0 2021-10-26 [2] Bioconductor
## xfun 0.40 2023-08-09 [1] CRAN (R 4.1.3)
## XMAS2 * 2.2.0 2023-11-30 [1] local
## XML 3.99-0.14 2023-03-19 [2] CRAN (R 4.1.2)
## xml2 1.3.5 2023-07-06 [2] CRAN (R 4.1.3)
## xtable 1.8-4 2019-04-21 [2] CRAN (R 4.1.0)
## XVector 0.34.0 2021-10-26 [2] Bioconductor
## yaml 2.3.7 2023-01-23 [2] CRAN (R 4.1.2)
## zlibbioc 1.40.0 2021-10-26 [2] Bioconductor
## zoo 1.8-12 2023-04-13 [2] CRAN (R 4.1.2)
##
## [1] /Users/zouhua/Library/R/x86_64/4.1/library
## [2] /Library/Frameworks/R.framework/Versions/4.1/Resources/library
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────