XMAS 2.0 Tutorial
2023-11-30
Chapter 1 Introduction
The next generation sequencing (NGS) technology have flourished the microbial research. More and more uncultured microbiota have been sequenced and identified, and their roles on host also have been investigated. According to the sequenced DNA parts, there are several methods including Amplicon sequencing (16s rRNA) and Whole genome shotgun sequencing (Metagenomics) to obtain the bacteria.
Amplicon sequencing (16s rRNA): uses the V3/V4 of bacteria 16s rRNA to sequence the bacteria. The in-house 16s sequencing pipeline uses QIIME2 platform wrapped DADA2 algorithm to acquire the amplicon sequence variants (ASVs) which is the same meaning of operational taxonomic units (OTUs).
Whole genome shotgun sequencing (Metagenomics): could detect the whole DNA of the microbial community. The in-house metagenomic sequencing pipeline uses the metaphlan2 or metaphlan3 algorithm which is based on marker genes of microbiome to obtain the microbial profiles.
We provide the universal data analysis framework on microbial data for in-house use when you use XMAS 2.0, and some specific data analysis modules should be performed by yourself in a different way.
If you have no any background on microbiota data analysis, please go to the example of Chapter 15 to get familiar with the SOP of data analysis. Alternatively, you can also go to the following websites:
This tutorial requires that the users should have basic knowledge on R language and statistics. If you are new to microbial research, we recommend the Statistical Analysis of Microbiome Data with R (Xia et al. 2018) as guide book.
We suggest users using local laptop because our own server have not deployed XMAS 2.0 package yet. In addition, installing the following software before using XMAS 2.0.
R 3.6.3 or later release Download link.
Rstudio Desktop Download link.
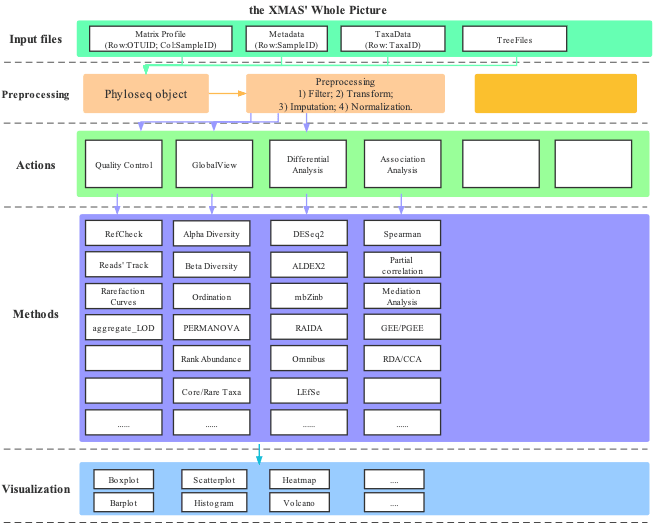
1.2 Input data
Note the data pre-processing requirements before analysing data with XMAS 2.0:
Types of data. Different types of sequencing data can be explored and integrated with XMAS 2.0. Our methods can handle not only 16s sequencing data by DADA2 but also metagenomic sequencing data by Metaphlan software. All the data should from the in-house pipeline.
dada2 result from standardized_analytics_workflow_R_function.
/home/xuxiaomin/project/standardized_analytics_workflow_R_function/demo_data/16S/process/xdada2/dada2_res.rds
/home/xuxiaomin/project/standardized_analytics_workflow_R_function/demo_data/16S/process/fasta2tree/tree.nwk
/home/xuxiaomin/project/standardized_analytics_workflow_R_function/demo_data/16S/metadata.txt
The result of the in-house Metaphlan2/3 pipeline.
/home/xuxiaomin/project/standardized_analytics_workflow_R_function/demo_data/MGS/metaphlan2_merged.tsv
/home/xuxiaomin/project/standardized_analytics_workflow_R_function/demo_data/MGS/metadata.txt
Transformation. The package handles transformation by log with 10.
Normalization. The package handles Normalization by providing many appropriate normalization methods.
Filtering or Trimming. While XMAS 2.0 methods can handle large data sets, we recommend pre-filtering the data to less than the threshold by features or samples. Such step aims to lessen the computational time and reduce the false positive rate during the downstream data analysis.
Data format. Our methods use phyloseq object as inputdata format. Therefore, the phyloseq object must be generated by our own methods (otu_table, sample_data or taxa_table).
Multiple groups’ comparison. In the current version of XMAS 2.0, it doesn’t provide the methods for Multiple groups’ comparison.
1.3 Methods
Generalized UniFrac Distance Metrics: Associating microbiome composition with environmental covariates using generalized UniFrac distances (J. Chen et al. 2012).
PERMANOVA: Permutational multivariate analysis of variance (PERMANOVA) (Anderson 2014).
Mantel Test: The detection of disease clustering and a generalized regression approach (Mantel 1967).
Multi-response permutation procedures (MRPP): The application of multivariate permutation methods based on distance functions in the earth sciences (Mielke Jr 1991).
ALDEx2: Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis (Fernandes et al. 2014).
limma: voom: Precision weights unlock linear model analysis tools for RNA-seq read counts (Law et al. 2014).
mbzinb: An omnibus test for differential distribution analysis of microbiome sequencing data (J. Chen et al. 2018).
RAIDA: A robust approach for identifying differentially abundant features in metagenomic samples (Sohn, Du, and An 2015).
LEfSe: Metagenomic biomarker discovery and explanation (Segata et al. 2011).
MetagenomeSeq: Differential abundance analysis for microbial marker-gene surveys (Paulson et al. 2013).
DESeq2: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 (Love, Huber, and Anders 2014).
edgeR: A scaling normalization method for differential expression analysis of RNA-seq data (Robinson and Oshlack 2010).
Analysis of composition of microbiomes (ANCOM): Analysis of composition of microbiomes: a novel method for studying microbial composition”, Microbial Ecology in Health (Mandal et al. 2015).
Corncob: Modeling microbial abundances and dysbiosis with beta-binomial regression”, Microbial Ecology in Health (Martin, Witten, and Willis 2020).
Maaslin2: Multivariable association discovery in population-scale meta-omics studies (Mallick et al. 2021).
LOCOM: A logistic regression model for testing differential abundance in compositional microbiome data with false discovery rate control (Hu, Satten, and Hu 2022).
1.4 Outline of this Tutorial
This tutorial was organized according to the XMAS 2.0 functions.
In the beginning two chapters, we specially provided the basic requirements of bioinformatics and overview on microbiota, and the installation of XMAS.
In Chapter 3, we convert the results from in-house pipeline into phyloseq-class object for downstream data analysis.
In Chapter 4 provided the functions to evaluate the data quality.
In Chapter 5, we introduced the preprocessing methods on microbiota data before differential analysis.
In Chapter 6 provided the alpha diversity analysis.
In Chapter 7 provided the beta diversity analysis.
In Chapter 8 provided the microbiota composition analysis.
In Chapter 9 provided the core microbiota analysis.
In Chapter 10 provided the differential analysis.
In Chapter 11 provided some functions for visualization.
In Chapter 12 provided the association analysis.
In Chapter 13 showed the principals of the differential analysis methods.
In Chapter 14 provided the examples by using XMAS 2.0 on microbiota data.
1.5 Key publications
phyloseq object. (McMurdie and Holmes 2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data.
Generalized UniFrac Distance Metrics. (J. Chen et al. 2012) Associating microbiome composition with environmental covariates using generalized UniFrac distances.
Permutational Multivariate Analysis of Variance (PERMANOVA). (Anderson 2014) Permutational multivariate analysis of variance (PERMANOVA).
ALDEx2. (Fernandes et al. 2014) Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis.
1.6 Citation
Kindly cite by using citation("XMAS") if you think XAMS helps you. Alternative way is Tong B, Zou H (2022). XMAS: A set of tools for statistical analysis in metagenomics. R package version 2.1.7, <URL:https://gitlab.xbiome.com/Analytics/xmas/>.