Chapter 3 Functional Analysis of KEGG
This demo guides you how to analyze KEGG pathway profile and KO profile.
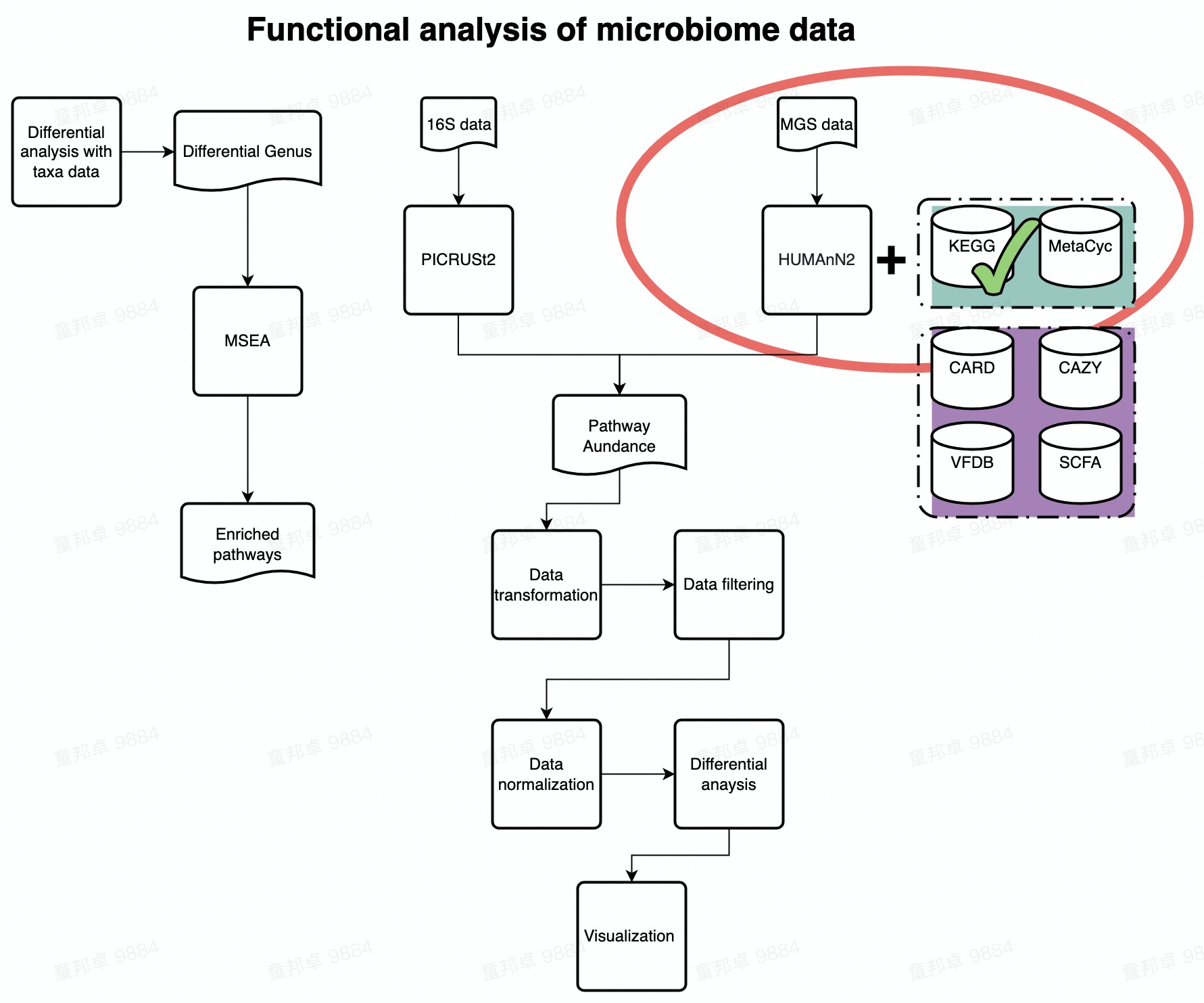
FlowChart_KEGG
3.1 Environment setup
3.2 Pipeline file processing (KEGG)
As described in the introduction chapter, a cohort containing 8 MGS samples of 4 patients from 2 groups in MAFLD project would be used as demo data in this tutorial.
Note: Pathway abundance can be calculated in many ways (median/mean/avarage_top_half value of KO profiles), in this tutorial we use “median” as example.
3.2.1 Remove description in KEGG map profile
Here we need to remove the description column of kegg map profile of 8 samples in Bash. But if you have removed the column data already, please skip this chunk and jump to Merge KEGG map profile of samples.
for i in /share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/PipelineOutput/*/humann2_post_kegg/*_pathway_abundance_median_des.tsv; do awk -F '\t' '{print $1"\t"$2}' $i > $i.tmp.tsv ;done3.2.2 Merge KEGG map profile of samples
Here we need to merge all data from 8 samples into one profile table with the merge script from Humann2 in Bash. But if you have merged the sample data already, please skip this chunk and jump to Data loading.
3.3 Data loading (KEGG)
pathway_profile_tbl <- read.csv('/share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/Kegg/merged_kegg_profile.tsv', header = TRUE, comment.char = '', stringsAsFactors = FALSE , na.strings = '', sep='\t', check.names = FALSE)
colnames(pathway_profile_tbl)[1] <- 'PathwayID'
## Remove suffix in sample SeqIDs
colnames(pathway_profile_tbl) %<>% str_remove_all('_.+')
## Remove prefix in map IDs
pathway_profile_tbl$PathwayID %<>% str_remove_all('map')
## We can map pathway ID to pathway names with the mapping file in minpath.
## But it is not suggested to do the mapping in this step, because pathway names contain species characters, but we read in mapping file here anyways.
ID2Name_mapping_tbl <- read.table('/share/work/runtime/softwares/MinPath/data/KEGG-pathway.txt', header = FALSE, na.strings = '', comment.char = '', sep = '\t', colClasses = c('V1' = 'character'))
colnames(ID2Name_mapping_tbl) <- c('PathwayID', 'Label')
## Next we read in metadata (Group information)
metadata <- read.table('/share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/metadata.xls', check.names = FALSE, header = TRUE)
metadata %<>% mutate(SeqID2 = SeqID) %>% column_to_rownames('SeqID2') %>% as.data.frame()3.4 Data preprocessing (KEGG)
3.4.1 Remove 0 abundance maps
Remove the pathways whose abundance are 0 across all samples. If we use harmonic mean or median to calcualte pathway abundance from KOs, the pathway abundance may be 0, we therefore need to remove those pathways having 0 abundance across all samples.
## If we use harmonic mean or median to calcualte pathway abundance from KOs, the pathway abundance may be 0, we therefore need to remove those pathways having 0 abundance across all samples.
pathway_profile_tbl %<>% column_to_rownames('PathwayID') %>% dplyr::filter(rowSums(.) > 0)
dim(pathway_profile_tbl)## [1] 63 83.4.2 Transforming data (KEGG)
Note: Run Remove 0 abundance maps before running this chunk!
## In this chunk, we use TSS (Total sum scaling) to eliminate the influence of sequencing depth on samples. After re-scaling, we can apply DA to samples.
rescaled_pathway_profile_tbl <- pathway_profile_tbl %>% apply(., 2, function(x) x/sum(x)) %>% as.data.frame()
dim(rescaled_pathway_profile_tbl)## [1] 63 83.4.3 Aggregate low abundance data
In this chunck, we aggregate low abundance features to one row.
1e-12 is an empirical threshold fot filtering low abundance feature. According to published paper Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition, pathway with top 50% mean abundance and top 50% variance are left. But in MaAsLin2,pathway with abundance less than 10-10 are filtered by default.
Note: Run Transforming data before running this chunk!
filtered_pathway_RA_profile_tbl <- aggregate_low_abundance(input_data = rescaled_pathway_profile_tbl,
threshold = 1e-12) ## threshold should be modified based on your on study
dim(filtered_pathway_RA_profile_tbl)## [1] 63 83.5 Standard Analysis (KEGG)
Note: All chunks in Data preprocessing should be excuted before doing analysis in this chunk.
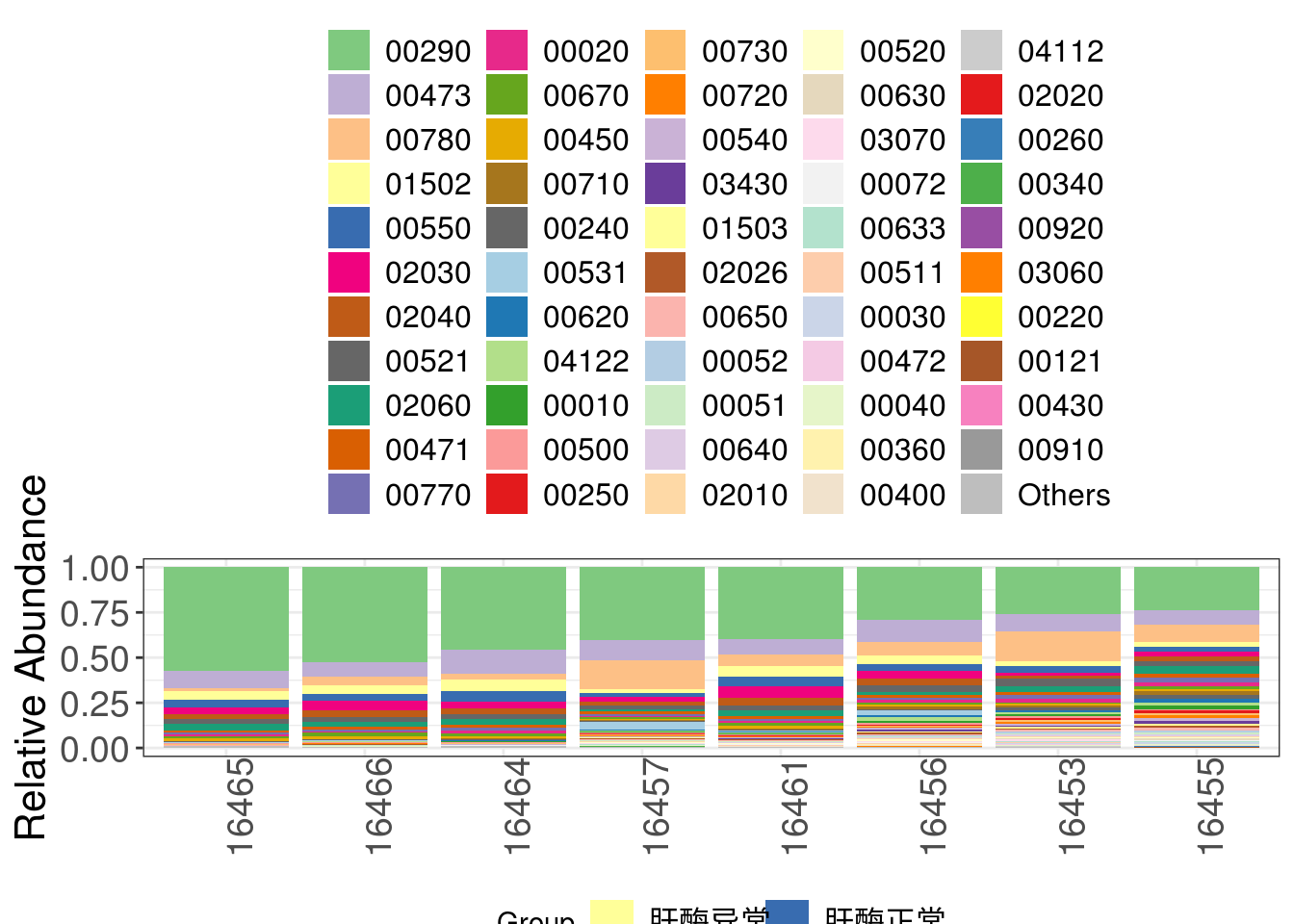
3.5.1 Compositional barplot
Generate compositional barplot
## In this chunk, we construct stacked pathway barplot to depict the pathway composition of samples, we use function plot_stacked_bar from xviz to plot.
## In case there are too much entries, we use parameter "collapse" in plot_stacked_bar function to integrate entries whose abundance are below given threshold into "Others".
## Note: make sure your graph is long enough to show the group information.
pathway_stacked_barplot <- xviz::plot_stacked_bar(otu_table = filtered_pathway_RA_profile_tbl %>% t() %>% as.data.frame(),
metadata = metadata, collapse = 1e-3, feature = 'Group')Plot compositional barplot
3.5.2 Beta diversity
## In this chunk, we inherit the concept of Beta diversity of microbial taxa data and apply it to pathway data to explore the similarity between samples.
## construct phyloseq for beta diversity analysis
phy <- phyloseq(otu_table(filtered_pathway_RA_profile_tbl, taxa_are_rows = TRUE),
sample_data(metadata))
## PCOA plot with bray_curtis distance
PCOA_plot <- xviz::plot_beta_diversity(phyloseq = phy,
feature = 'Group',
method = 'bray',
label = TRUE)
print(PCOA_plot)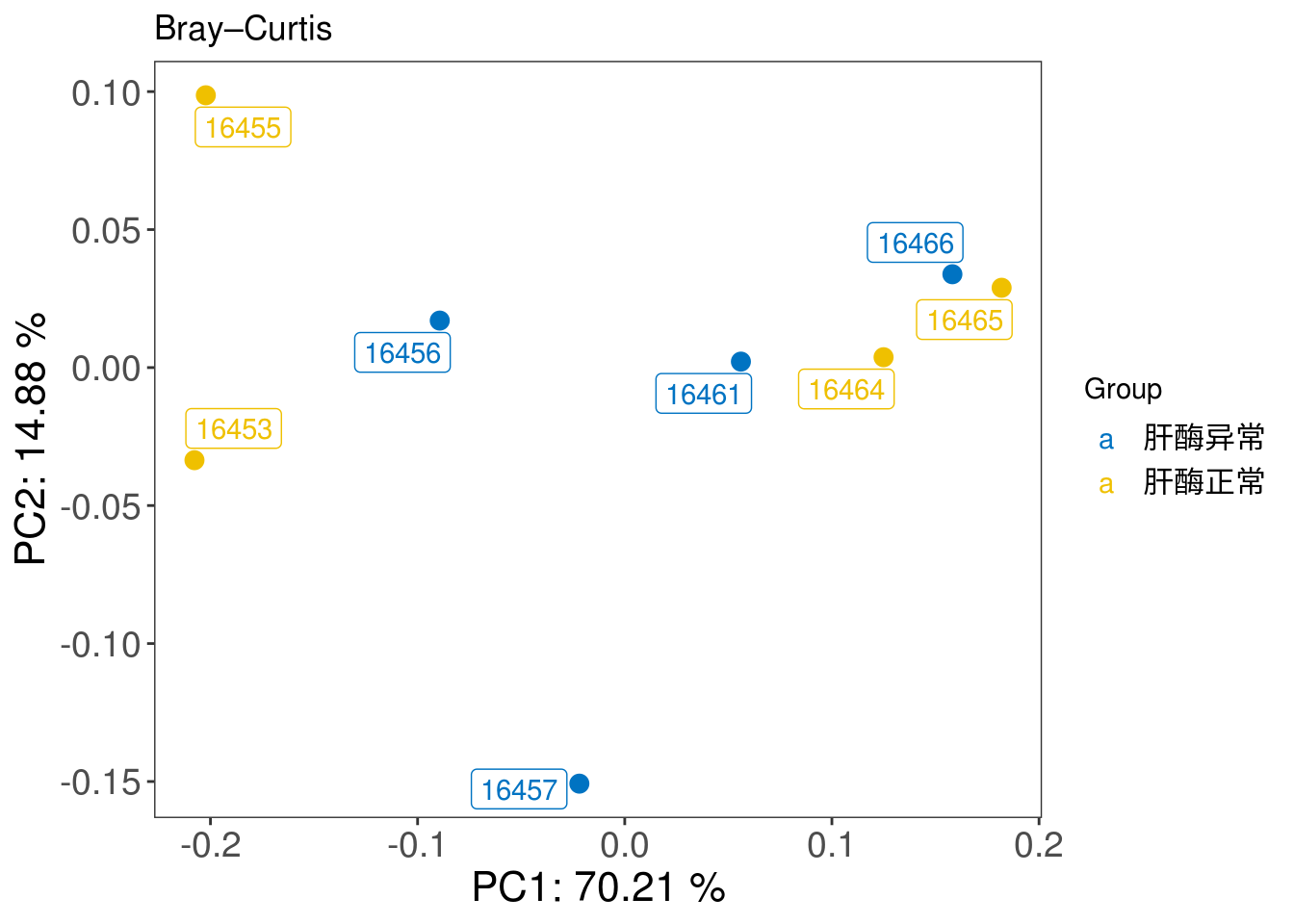
## PERMANOVA test & beta dispersion test
## We use PERMANOVA test to check the differences of function composition among different groups. Additionally, we also take homogeneity of group variance into consideration.
dispersion_permanova_res <- run_permanova_betadisp(physeq = phy,
vars = 'Group'
)
dispersion_permanova_res## $betadisp_res
## variable p_value analysis
## 1 Group 0.109 beta_dispersion_permutation999
##
## $permanova_res
## variable p_value R2 analysis
## 1 Group 0.622 0.08514936 permanova_permutation9993.6 Differential analysis (KEGG)
Note: All chunks in Data preprocessing should be executed before doing analysis in this chunk.
3.6.1 Filter low prevalence pathway (KEGG)
Low prevalence pathways are pathways only occur in minor samples. In this chunk, we would remove pathways apperaing in less than max(2 , 5% of samples) from data set before doing analysis. **If you have filtered the low prevalence pathways please jump to DA with Logistic Regression for differential analysis.
filtered_pathway_RA_profile_tbl <- filter_prevalence(otu_table = filtered_pathway_RA_profile_tbl,
threshold = 0.05,
taxa_are_rows = TRUE
)
dim(filtered_pathway_RA_profile_tbl)## [1] 61 83.6.2 DA with Logistic Regression
In this chunk, you would be using logistic regression model to find pathways that are significantly enriched in certain group.
Note: Remeber to Filter low prevalence pathway before running this chunk.
## Due to the nature of compositional data, we cannot apply linear models to compositional data directly.
## Transformation of relative abundance data should be carried out before feeding the data to LM.
## Here, we add a very small value (1e-12) to the pathway profile table to avoid genrating NA during transformation, then use logit transformation to transform data. And eventually we apply LM to the transformed data
## Adding small value to the profile table (The value is arbitrary).
DA_pathway_RA_profile_tbl <- filtered_pathway_RA_profile_tbl + 1e-12
## Reshape profile data table and use logit transformation.
DA_pathway_RA_profile_tbl <- DA_pathway_RA_profile_tbl %>% t() %>% as.data.frame() %>% rownames_to_column('SeqID') %>% as.data.frame()
DA_metadata <- metadata %>% as.data.frame()
## Reshape dataframe into long table
DA_input <- merge(DA_pathway_RA_profile_tbl, DA_metadata, by='SeqID') %>% reshape2::melt(value.name = 'RA',
variable.name = 'PathwayIDs')
## Logit transformation
DA_input %<>% mutate(RA_logit = log(RA/(1-RA)))
## Fit data to LM
## Loop over each pathway in two groups
LM_res <- DA_input %>% split(.$PathwayIDs) %>% lapply(., function(x){
gml_res_summary <- lm(data = x, formula = RA_logit ~ Group) %>% summary() %>% .$coefficients %>% as.matrix() %>% as.data.frame() %>% rownames_to_column(var = "Factors")
}
)
## Merge all result in one table
LM_res <- LM_res %>% data.table::rbindlist(idcol = "PathwayID") %>% filter(Factors != "(Intercept)")
## Adjust p value using p.adjust function from stats package, you could choose different adjust method.
LM_res %<>% mutate(adjust.p = stats::p.adjust(.$`Pr(>|t|)`,
method = 'BH'))
## Calculate effect size (Odds ratio) of each feature
LM_res %<>% mutate(OR = exp(Estimate)) %>% as.data.frame()
## Add label to pathwayIDs
LM_res %<>% merge(., ID2Name_mapping_tbl, by = 'PathwayID')3.6.3 Show DA result with volcano plot
## Plot volcano plot to show effect size (x-axis) and p value (y-axis) of pathways.
## Here we only tend to hightlight pathways that satisfy adjust.p < 0.614 and (OR < 0.2 | OR > 0.5) at the same time.
## You can nevertheless choose different threshold accroding to your own data.
volcano_plot <- LM_res %>% mutate(p.adj.log = -log10(adjust.p), log10OR = log10(OR)) %>%
ggplot(aes(x = log10OR, y = p.adj.log)) +
geom_point(size = 0.5) +
geom_point(size = 0.5, color = "red", data = . %>% filter(adjust.p < 0.614 & (OR < 0.2 | OR > 0.5))) +
#geom_text_repel(size = 6/.pt, aes(label = Label), data = . %>% filter(adjust.p<0.614 & OR<0.2)) +
geom_vline(xintercept = log10(c(0.05, 0.1, 0.2, 0.5, 1, 2)), size = 0.05, color = "grey") +
geom_hline(yintercept = -log10(c(0.7)), size = 0.05, color = "grey") +
theme(aspect.ratio = 1,
panel.grid.minor = element_blank(),
panel.grid.major.x = element_blank()
) +
labs(x = "Estimated Odds Ratio", y = "FDR adjusted p-values(-log10)")
print(volcano_plot)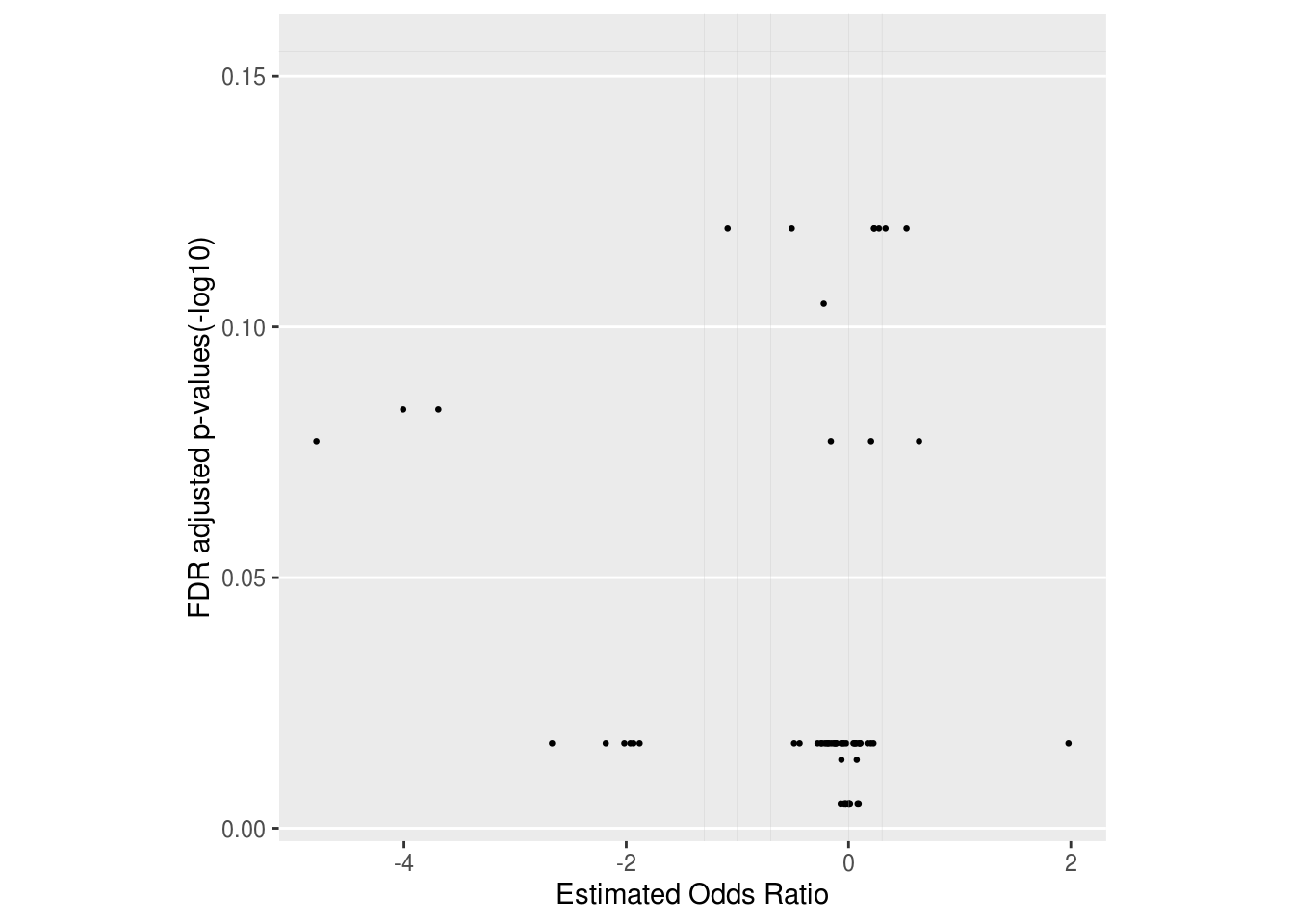
3.6.4 Show DA result with pathview
In this chunk, we use pathview to visualize enriched KOs in differential pathways.
Step1. First we extract all KOs in differential pathways obtained from last chunk with mapping file: /share/work/runtime/softwares/MinPath/data/KEGG-mapping.txt.
Step2. We find differential KO in two groups using the same LM method in previous chunk.
Step3. Visualize differential KO in differential pathways with pathview.
In order to find differential KO in two groups, we need to merge all profiles of all samples
3.6.4.1 Merge KO profiles
/share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/PipelineOutput/*/humann2_post_kegg/16466_filter_unstratified_with_des.kegg3.6.4.1.1 Remove description in KO profile
Here we need to remove the description column of KO profile of 8 samples in Bash. But if you have removed the column data already, please skip this chunk and jump to Read merged KO profile.
for i in /share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/PipelineOutput/*/humann2_post_kegg/*filter_unstratified_with_des.kegg; do awk -F '\t' '{print $1"\t"$2}' $i > $i.tmp.tsv ;done3.6.4.1.2 Merge KO profile of all sample
Here we need to merge all KO data from 8 samples into one profile table with the merge script from Humann2 in Bash. But if you have merged the sample data already, please skip this chunk and jump to Read merged KO profile.
3.6.4.1.3 Remove tmp KO tsv files
Remove tmp tsv files created above
3.6.4.2 Read merged KO profile
## Read in KO profile, since we've already removed UNMAPPED and re-scaled KO profile in Functional_Analysis_Kegg_Pathway_preprocess.Rmd, we only need to filer low_abundance and low-prevalenece KOs here.
KO_profile <- read.table('/share/projects/SOP/Functional_Analysis/Tongbangzhuo/Demodata/Kegg/merged_ko_profile.tsv', header = TRUE, check.names = FALSE)
colnames(KO_profile) %<>% str_remove('_.+')3.6.4.3 Preprocess merged KO profile
## Rescale KO profile
KO_profile %<>% column_to_rownames('ID') %>% apply(., 2, function(x) x/sum(x)) %>% as.data.frame() %>% rownames_to_column('KOs')
## Filter low abundance KOs
filtered_KO_profile <- KO_profile %>% column_to_rownames('KOs') %>% aggregate_low_abundance(input_data = .,
threshold = 1e-12)
## Filter low-prevalenece KOs
filtered_KO_profile <- filter_prevalence(otu_table = filtered_KO_profile,
threshold = 0.05,
taxa_are_rows = TRUE
)3.6.4.4 Find differential KO for each differential Kegg map
## Read in KO-Pathway mapping file
KO2Pathway_mapping <- read.table('/share/work/runtime/softwares/MinPath/data/KEGG-mapping.txt', colClasses = c('V1' = 'character'))
## Loop over differential pathways
Diff_pathway <- LM_res %>% filter(`Pr(>|t|)` < .05) %>% .$PathwayID %>% as.vector()
for (pathway in Diff_pathway){
## Create a list to store the log2fc of each KO in each kegg pathway
log2fc_list <- list()
## Extract all KOs in differential pathway
KO_ID_in_mapping <- KO2Pathway_mapping %>% filter(V1 == pathway) %>% .$V2
## Some KOs are not included in mapping file, here we take intersection of KO ID in mapping file and avaiable KO ID in KO profile
KO_ID <- intersect(KO_ID_in_mapping, rownames(filtered_KO_profile))
tmp_KO_profile <- filtered_KO_profile[KO_ID,]
tmp_KO_meta <- DA_metadata
## re-format tmp KO profile table
KO_DA_input <- merge(tmp_KO_profile %>% t() %>% as.data.frame() %>% rownames_to_column('SeqID'),
tmp_KO_meta,
by = 'SeqID') %>% reshape2::melt(variable.name = 'KO',
value.name = 'RA') %>% mutate(RA = RA + 1e-12) %>% mutate(RA_logit = log(RA/(1-RA)))
## DA using LM
KO_LM_res <- KO_DA_input %>% split(.$KO) %>% lapply(., function(x){
KO_gml_res_summary <- lm(data = x, formula = RA_logit ~ Group) %>% summary() %>% .$coefficients %>% as.matrix() %>% as.data.frame() %>% rownames_to_column(var = "Factors")
}
)
## Merge result of all tested KOs
KO_LM_res %<>% data.table::rbindlist(idcol = "KOID") %>% filter(Factors != "(Intercept)")
## Adjust p value using p.adjust function from stats package, you could choose different adjust method.
KO_LM_res %<>% mutate(adjust.p = stats::p.adjust(.$`Pr(>|t|)`,
method = 'BH')) %>% as.data.frame()
## Filter significant differential KOs with p-value, we use pvalue < 0.05 as filtering criteria here, choose your own standard for filtering.
Sig_KO <- KO_LM_res %>% filter(`Pr(>|t|)` < 0.05) %>% .$KOID
## Calculate Log2FC of each significant KO in two groups
for (ko in Sig_KO){
## Add a small value to avoid 0
Sig_KO_profile <- tmp_KO_profile[ko, ] + 1e-12
## calculate median for each KO in two groups
Sig_KO_log2fc <- merge(Sig_KO_profile %>% t() %>% as.data.frame() %>% rownames_to_column('SeqID'),
tmp_KO_meta,
by = 'SeqID') %>% column_to_rownames('SeqID') %>% group_by(Group) %>% summarize(median = median(!!as.symbol(ko))) %>% as.data.frame()
## Here we extact the median value for each group separatly
GroupA_median <- Sig_KO_log2fc %>% column_to_rownames('Group') %>% .['A', 'median']
GroupB_median <- Sig_KO_log2fc %>% column_to_rownames('Group') %>% .['B', 'median']
## Calculate log2FC for each KO and save the value in list
## Please be clear which group is divided by the other, it decides the direction of your log2fc
## Positive log2fc value would be highlighted in red in pathview result while negative log2fc value would be highlighted in green
log2fc_list[[ko]] <- log2(GroupA_median/GroupB_median)
}
## converst list to data frame and use it as input for pathview
input <- unlist(log2fc_list) %>% as.data.frame()
## Visualization
pathview(input, pathway.id = pathway, species = "ko", out.suffix= 'PathView')
}
## Knit Pathview picture to RMD
graph_list <- system('ls *.PathView.png', intern = TRUE)
## In the graph(s) below, green KO are those KOs enriched in GroupB, red KO are those KOs enriched in GroupA
knitr::include_graphics(graph_list)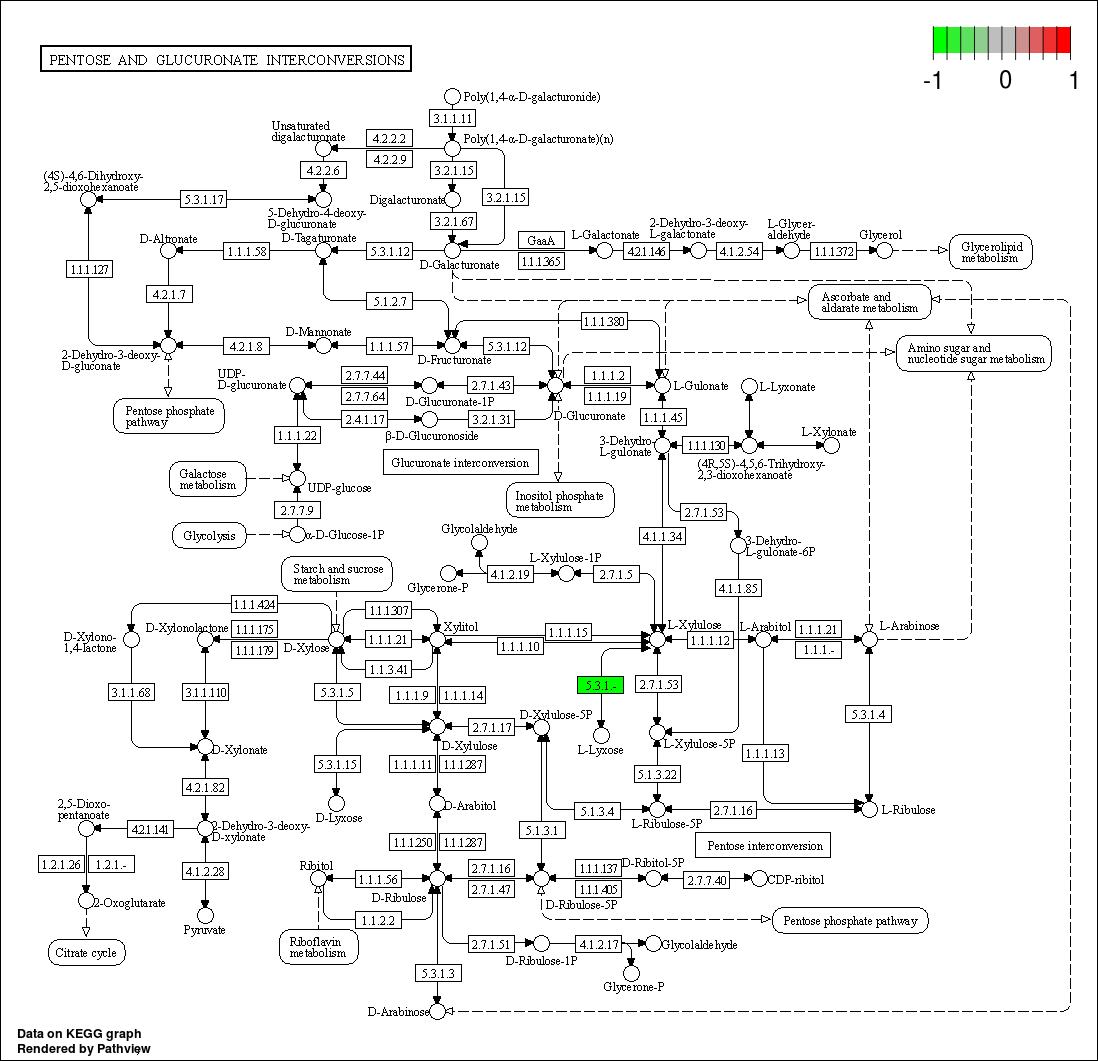

3.7 Session information (KEGG)
## ─ Session info ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 3.6.3 (2020-02-29)
## os Ubuntu 16.04.7 LTS
## system x86_64, linux-gnu
## ui RStudio
## language (EN)
## collate en_IN.UTF-8
## ctype en_IN.UTF-8
## tz Asia/Hong_Kong
## date 2022-11-09
## rstudio 1.1.419 (server)
## pandoc 2.7.3 @ /usr/bin/ (via rmarkdown)
##
## ─ Packages ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## ! package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [1] CRAN (R 3.6.3)
## ade4 1.7-17 2021-06-17 [1] CRAN (R 3.6.3)
## ALDEx2 * 1.18.0 2019-10-29 [1] Bioconductor
## annotate 1.64.0 2019-10-29 [1] Bioconductor
## AnnotationDbi * 1.58.0 2022-04-26 [1] Bioconductor
## ape 5.5 2021-04-25 [1] CRAN (R 3.6.3)
## assertthat 0.2.1 2019-03-21 [2] CRAN (R 3.6.3)
## backports 1.4.1 2021-12-13 [1] CRAN (R 3.6.3)
## base64enc 0.1-3 2015-07-28 [2] CRAN (R 3.6.3)
## bayesm 3.1-4 2019-10-15 [1] CRAN (R 3.6.3)
## biglm 0.9-2.1 2020-11-27 [1] CRAN (R 3.6.3)
## Biobase * 2.46.0 2019-10-29 [2] Bioconductor
## BiocGenerics * 0.32.0 2019-10-29 [2] Bioconductor
## BiocParallel * 1.20.1 2019-12-21 [2] Bioconductor
## biomformat 1.14.0 2019-10-29 [1] Bioconductor
## Biostrings 2.54.0 2019-10-29 [1] Bioconductor
## bit 4.0.4 2020-08-04 [1] CRAN (R 3.6.3)
## bit64 4.0.5 2020-08-30 [1] CRAN (R 3.6.3)
## bitops 1.0-7 2021-04-24 [1] CRAN (R 3.6.3)
## blob 1.2.2 2021-07-23 [1] CRAN (R 3.6.3)
## bookdown 0.24 2021-09-02 [1] CRAN (R 3.6.3)
## brio 1.1.3 2021-11-30 [2] CRAN (R 3.6.3)
## broom 0.7.12 2022-01-28 [1] CRAN (R 3.6.3)
## bslib 0.3.1 2021-10-06 [1] CRAN (R 3.6.3)
## cachem 1.0.5 2021-05-15 [1] CRAN (R 3.6.3)
## callr 3.7.0 2021-04-20 [2] CRAN (R 3.6.3)
## car 3.0-12 2021-11-06 [1] CRAN (R 3.6.3)
## carData 3.0-4 2020-05-22 [1] CRAN (R 3.6.3)
## caTools 1.18.2 2021-03-28 [1] CRAN (R 3.6.3)
## cellranger 1.1.0 2016-07-27 [1] CRAN (R 3.6.3)
## checkmate 2.0.0 2020-02-06 [1] CRAN (R 3.6.3)
## circlize * 0.4.13 2021-06-09 [1] CRAN (R 3.6.3)
## cli 3.1.0 2021-10-27 [1] CRAN (R 3.6.3)
## clue 0.3-59 2021-04-16 [1] CRAN (R 3.6.3)
## cluster 2.1.0 2019-06-19 [2] CRAN (R 3.6.3)
## coda 0.19-4 2020-09-30 [1] CRAN (R 3.6.3)
## codetools 0.2-16 2018-12-24 [2] CRAN (R 3.6.3)
## coin 1.4-2 2021-10-08 [1] CRAN (R 3.6.3)
## colorspace 2.0-2 2021-06-24 [1] CRAN (R 3.6.3)
## ComplexHeatmap * 2.2.0 2019-10-29 [1] Bioconductor
## compositions 2.0-2 2021-07-14 [1] CRAN (R 3.6.3)
## cowplot * 1.1.1 2020-12-30 [1] CRAN (R 3.6.3)
## crayon 1.5.0 2022-02-14 [1] CRAN (R 3.6.3)
## curl 4.3.2 2021-06-23 [2] CRAN (R 3.6.3)
## dada2 * 1.14.1 2020-02-22 [1] Bioconductor
## data.table * 1.14.0 2021-02-21 [1] CRAN (R 3.6.3)
## DBI 1.1.1 2021-01-15 [1] CRAN (R 3.6.3)
## dbplyr 2.1.1 2021-04-06 [1] CRAN (R 3.6.3)
## DelayedArray * 0.12.3 2020-04-09 [2] Bioconductor
## DelayedMatrixStats 1.8.0 2019-10-29 [1] Bioconductor
## DEoptimR 1.0-9 2021-05-24 [1] CRAN (R 3.6.3)
## desc 1.4.1 2022-03-06 [2] CRAN (R 3.6.3)
## DESeq2 * 1.26.0 2019-10-29 [1] Bioconductor
## devtools 2.4.3 2021-11-30 [1] CRAN (R 3.6.3)
## digest 0.6.29 2021-12-01 [1] CRAN (R 3.6.3)
## dplyr * 1.0.6 2021-05-05 [1] CRAN (R 3.6.3)
## edgeR 3.28.1 2020-02-26 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [1] CRAN (R 3.6.3)
## EnhancedVolcano * 1.4.0 2019-10-29 [1] Bioconductor
## enrichR * 3.0 2021-02-02 [1] CRAN (R 3.6.3)
## evaluate 0.15 2022-02-18 [2] CRAN (R 3.6.3)
## fansi 1.0.2 2022-01-14 [1] CRAN (R 3.6.3)
## farver 2.1.0 2021-02-28 [2] CRAN (R 3.6.3)
## fastmap 1.1.0 2021-01-25 [1] CRAN (R 3.6.3)
## fdrtool 1.2.17 2021-11-13 [1] CRAN (R 3.6.3)
## forcats * 0.5.1 2021-01-27 [1] CRAN (R 3.6.3)
## foreach 1.5.2 2022-02-02 [2] CRAN (R 3.6.3)
## foreign 0.8-75 2020-01-20 [2] CRAN (R 3.6.3)
## formatR 1.12 2022-03-31 [2] CRAN (R 3.6.3)
## Formula 1.2-4 2020-10-16 [1] CRAN (R 3.6.3)
## fs 1.5.2 2021-12-08 [1] CRAN (R 3.6.3)
## futile.logger 1.4.3 2016-07-10 [2] CRAN (R 3.6.3)
## futile.options 1.0.1 2018-04-20 [2] CRAN (R 3.6.3)
## genefilter 1.68.0 2019-10-29 [1] Bioconductor
## geneplotter 1.64.0 2019-10-29 [1] Bioconductor
## generics 0.1.2 2022-01-31 [1] CRAN (R 3.6.3)
## GenomeInfoDb * 1.22.1 2020-03-27 [2] Bioconductor
## GenomeInfoDbData 1.2.2 2020-08-24 [2] Bioconductor
## GenomicAlignments 1.22.1 2019-11-12 [1] Bioconductor
## GenomicRanges * 1.38.0 2019-10-29 [2] Bioconductor
## getopt 1.20.3 2019-03-22 [1] CRAN (R 3.6.3)
## GetoptLong 1.0.5 2020-12-15 [1] CRAN (R 3.6.3)
## GGally * 2.1.2 2021-06-21 [1] CRAN (R 3.6.3)
## ggbipart * 0.1.2 2022-07-20 [1] Github (pedroj/bipartite_plots@162f577)
## ggExtra * 0.9 2019-08-27 [1] CRAN (R 3.6.3)
## ggplot2 * 3.3.5 2021-06-25 [1] CRAN (R 3.6.3)
## ggpubr * 0.4.0 2020-06-27 [1] CRAN (R 3.6.3)
## ggrepel * 0.9.1 2021-01-15 [2] CRAN (R 3.6.3)
## ggsci * 2.9 2018-05-14 [1] CRAN (R 3.6.3)
## ggsignif 0.6.3 2021-09-09 [1] CRAN (R 3.6.3)
## glmnet 4.1-2 2021-06-24 [1] CRAN (R 3.6.3)
## GlobalOptions 0.1.2 2020-06-10 [1] CRAN (R 3.6.3)
## glue 1.6.1 2022-01-22 [1] CRAN (R 3.6.3)
## GMPR 0.1.3 2021-05-17 [1] local
## gplots 3.1.1 2020-11-28 [1] CRAN (R 3.6.3)
## graph 1.64.0 2019-10-29 [1] Bioconductor
## gridExtra 2.3 2017-09-09 [2] CRAN (R 3.6.3)
## gtable 0.3.0 2019-03-25 [2] CRAN (R 3.6.3)
## gtools 3.9.2 2021-06-06 [1] CRAN (R 3.6.3)
## haven 2.4.1 2021-04-23 [1] CRAN (R 3.6.3)
## highr 0.9 2021-04-16 [1] CRAN (R 3.6.3)
## Hmisc 4.5-0 2021-02-28 [1] CRAN (R 3.6.3)
## hms 1.1.1 2021-09-26 [1] CRAN (R 3.6.3)
## htmlTable 2.3.0 2021-10-12 [1] CRAN (R 3.6.3)
## htmltools 0.5.2 2021-08-25 [1] CRAN (R 3.6.3)
## htmlwidgets 1.5.4 2021-09-08 [2] CRAN (R 3.6.3)
## httpuv 1.6.1 2021-05-07 [1] CRAN (R 3.6.3)
## httr 1.4.3 2022-05-04 [2] CRAN (R 3.6.3)
## hwriter 1.3.2 2014-09-10 [1] CRAN (R 3.6.3)
## igraph 1.3.1 2022-04-20 [2] CRAN (R 3.6.3)
## IHW 1.14.0 2019-10-29 [1] Bioconductor
## IRanges * 2.20.2 2020-01-13 [2] Bioconductor
## iterators 1.0.14 2022-02-05 [2] CRAN (R 3.6.3)
## jpeg 0.1-9 2021-07-24 [1] CRAN (R 3.6.3)
## jquerylib 0.1.4 2021-04-26 [1] CRAN (R 3.6.3)
## jsonlite 1.8.0 2022-02-22 [2] CRAN (R 3.6.3)
## KEGGgraph 1.46.0 2019-10-29 [1] Bioconductor
## KEGGREST 1.26.1 2019-11-06 [1] Bioconductor
## KernSmooth 2.23-16 2019-10-15 [2] CRAN (R 3.6.3)
## knitr 1.36 2021-09-29 [1] CRAN (R 3.6.3)
## labeling 0.4.2 2020-10-20 [2] CRAN (R 3.6.3)
## lambda.r 1.2.4 2019-09-18 [2] CRAN (R 3.6.3)
## later 1.3.0 2021-08-18 [2] CRAN (R 3.6.3)
## lattice * 0.20-38 2018-11-04 [2] CRAN (R 3.6.3)
## latticeExtra 0.6-29 2019-12-19 [1] CRAN (R 3.6.3)
## lazyeval 0.2.2 2019-03-15 [2] CRAN (R 3.6.3)
## libcoin 1.0-9 2021-09-27 [1] CRAN (R 3.6.3)
## lifecycle 1.0.1 2021-09-24 [1] CRAN (R 3.6.3)
## limma 3.42.2 2020-02-03 [2] Bioconductor
## locfit 1.5-9.4 2020-03-25 [1] CRAN (R 3.6.3)
## lpsymphony 1.14.0 2019-10-29 [1] Bioconductor (R 3.6.3)
## lubridate 1.7.10 2021-02-26 [1] CRAN (R 3.6.3)
## Maaslin2 1.7.3 2022-03-23 [1] Github (biobakery/maaslin2@8d090e4)
## magrittr * 2.0.2 2022-01-26 [1] CRAN (R 3.6.3)
## MASS 7.3-54 2021-05-03 [1] CRAN (R 3.6.3)
## Matrix 1.3-4 2021-06-01 [1] CRAN (R 3.6.3)
## matrixStats * 0.60.0 2021-07-26 [1] CRAN (R 3.6.3)
## mbzinb 0.2 2021-06-23 [1] local
## memoise 2.0.1 2021-11-26 [2] CRAN (R 3.6.3)
## metagenomeSeq 1.28.2 2020-02-03 [1] Bioconductor
## metamicrobiomeR 1.1 2021-02-03 [1] local
## mgcv 1.8-31 2019-11-09 [2] CRAN (R 3.6.3)
## microbiome 1.8.0 2019-10-29 [1] Bioconductor
## mime 0.12 2021-09-28 [2] CRAN (R 3.6.3)
## miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 3.6.3)
## modelr 0.1.8 2020-05-19 [1] CRAN (R 3.6.3)
## modeltools 0.2-23 2020-03-05 [1] CRAN (R 3.6.3)
## multcomp 1.4-17 2021-04-29 [1] CRAN (R 3.6.3)
## multtest 2.42.0 2019-10-29 [2] Bioconductor
## munsell 0.5.0 2018-06-12 [2] CRAN (R 3.6.3)
## mvtnorm 1.1-3 2021-10-08 [1] CRAN (R 3.6.3)
## network * 1.17.1 2021-06-14 [1] CRAN (R 3.6.3)
## nlme 3.1-144 2020-02-06 [2] CRAN (R 3.6.3)
## nnet 7.3-12 2016-02-02 [2] CRAN (R 3.6.3)
## optparse 1.7.1 2021-10-08 [1] CRAN (R 3.6.3)
## org.Hs.eg.db * 3.10.0 2021-12-08 [1] Bioconductor
## pathview * 1.26.0 2019-10-29 [1] Bioconductor
## pcaPP 1.9-74 2021-04-23 [1] CRAN (R 3.6.3)
## permute * 0.9-5 2019-03-12 [1] CRAN (R 3.6.3)
## phyloseq * 1.30.0 2019-10-29 [1] Bioconductor
## pillar 1.7.0 2022-02-01 [1] CRAN (R 3.6.3)
## pkgbuild 1.3.1 2021-12-20 [2] CRAN (R 3.6.3)
## pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 3.6.3)
## pkgload 1.2.4 2021-11-30 [2] CRAN (R 3.6.3)
## plotly * 4.10.0 2021-10-09 [1] CRAN (R 3.6.3)
## plyr 1.8.7 2022-03-24 [2] CRAN (R 3.6.3)
## png 0.1-7 2013-12-03 [1] CRAN (R 3.6.3)
## prettyunits 1.1.1 2020-01-24 [2] CRAN (R 3.6.3)
## processx 3.5.3 2022-03-25 [2] CRAN (R 3.6.3)
## promises 1.2.0.1 2021-02-11 [2] CRAN (R 3.6.3)
## protoclust 1.6.3 2019-01-31 [1] CRAN (R 3.6.3)
## ps 1.7.0 2022-04-23 [2] CRAN (R 3.6.3)
## pscl 1.5.5 2020-03-07 [1] CRAN (R 3.6.3)
## purrr * 0.3.4 2020-04-17 [2] CRAN (R 3.6.3)
## qvalue 2.18.0 2019-10-29 [1] Bioconductor
## R6 2.5.1 2021-08-19 [1] CRAN (R 3.6.3)
## RAIDA 1.0 2021-06-23 [1] local
## ranacapa 0.1.0 2021-06-18 [1] Github (gauravsk/ranacapa@58c0cab)
## RColorBrewer * 1.1-3 2022-04-03 [2] CRAN (R 3.6.3)
## Rcpp * 1.0.7 2021-07-07 [1] CRAN (R 3.6.3)
## RcppParallel 5.1.4 2021-05-04 [1] CRAN (R 3.6.3)
## RCurl 1.98-1.6 2022-02-08 [2] CRAN (R 3.6.3)
## readr * 2.0.0 2021-07-20 [1] CRAN (R 3.6.3)
## readxl * 1.3.1 2019-03-13 [1] CRAN (R 3.6.3)
## remotes 2.4.2 2021-11-30 [1] CRAN (R 3.6.3)
## reprex 2.0.1 2021-08-05 [1] CRAN (R 3.6.3)
## reshape 0.8.9 2022-04-12 [1] CRAN (R 3.6.3)
## reshape2 * 1.4.4 2020-04-09 [2] CRAN (R 3.6.3)
## Rgraphviz 2.30.0 2019-10-29 [1] Bioconductor
## rhdf5 2.30.1 2019-11-26 [1] Bioconductor
## Rhdf5lib 1.8.0 2019-10-29 [1] Bioconductor
## rJava 1.0-5 2021-09-24 [1] CRAN (R 3.6.3)
## rjson 0.2.20 2018-06-08 [1] CRAN (R 3.6.3)
## R rlang 1.0.2 <NA> [2] <NA>
## rmarkdown 2.11 2021-09-14 [1] CRAN (R 3.6.3)
## robustbase 0.93-9 2021-09-27 [1] CRAN (R 3.6.3)
## rpart 4.1-15 2019-04-12 [2] CRAN (R 3.6.3)
## rprojroot 2.0.2 2020-11-15 [1] CRAN (R 3.6.3)
## Rsamtools 2.2.3 2020-02-23 [1] Bioconductor
## RSQLite 2.2.7 2021-04-22 [1] CRAN (R 3.6.3)
## rstatix 0.7.0 2021-02-13 [1] CRAN (R 3.6.3)
## rstudioapi 0.13 2020-11-12 [2] CRAN (R 3.6.3)
## Rtsne 0.15 2018-11-10 [1] CRAN (R 3.6.3)
## rvest 1.0.2 2021-10-16 [1] CRAN (R 3.6.3)
## S4Vectors * 0.24.4 2020-04-09 [2] Bioconductor
## sandwich 3.0-1 2021-05-18 [1] CRAN (R 3.6.3)
## sass 0.4.0 2021-05-12 [1] CRAN (R 3.6.3)
## scales 1.2.0 2022-04-13 [2] CRAN (R 3.6.3)
## seqinr * 4.2-8 2021-06-09 [1] CRAN (R 3.6.3)
## sessioninfo 1.2.2 2021-12-06 [2] CRAN (R 3.6.3)
## shape 1.4.6 2021-05-19 [1] CRAN (R 3.6.3)
## shiny 1.7.1 2021-10-02 [1] CRAN (R 3.6.3)
## ShortRead 1.44.3 2020-02-03 [1] Bioconductor
## slam 0.1-49 2021-11-17 [1] CRAN (R 3.6.3)
## statnet.common 4.5.0 2021-06-05 [1] CRAN (R 3.6.3)
## stringi 1.7.4 2021-08-25 [1] CRAN (R 3.6.3)
## stringr * 1.4.0 2019-02-10 [2] CRAN (R 3.6.3)
## SummarizedExperiment * 1.16.1 2019-12-19 [2] Bioconductor
## survival 3.1-8 2019-12-03 [2] CRAN (R 3.6.3)
## tensorA 0.36.2 2020-11-19 [1] CRAN (R 3.6.3)
## testthat 3.1.4 2022-04-26 [2] CRAN (R 3.6.3)
## textshape 1.7.3 2021-05-28 [1] CRAN (R 3.6.3)
## TH.data 1.1-0 2021-09-27 [1] CRAN (R 3.6.3)
## tibble * 3.1.6 2021-11-07 [1] CRAN (R 3.6.3)
## tidyr * 1.2.0 2022-02-01 [1] CRAN (R 3.6.3)
## tidyselect 1.1.1 2021-04-30 [1] CRAN (R 3.6.3)
## tidyverse * 1.3.1 2021-04-15 [1] CRAN (R 3.6.3)
## tzdb 0.2.0 2021-10-27 [1] CRAN (R 3.6.3)
## UpSetR 1.4.0 2019-05-22 [1] CRAN (R 3.6.3)
## usethis 2.1.6 2022-05-25 [2] CRAN (R 3.6.3)
## utf8 1.2.2 2021-07-24 [1] CRAN (R 3.6.3)
## vctrs 0.3.8 2021-04-29 [1] CRAN (R 3.6.3)
## vegan * 2.5-7 2020-11-28 [1] CRAN (R 3.6.3)
## VennDiagram 1.7.1 2021-12-02 [1] CRAN (R 3.6.3)
## viridisLite 0.4.0 2021-04-13 [2] CRAN (R 3.6.3)
## vroom 1.5.7 2021-11-30 [1] CRAN (R 3.6.3)
## wesanderson * 0.3.6.9000 2021-07-21 [1] Github (karthik/wesanderson@651c944)
## withr 2.4.3 2021-11-30 [1] CRAN (R 3.6.3)
## Wrench 1.4.0 2019-10-29 [1] Bioconductor
## xfun 0.23 2021-05-15 [1] CRAN (R 3.6.3)
## xlsx * 0.6.5 2020-11-10 [1] CRAN (R 3.6.3)
## xlsxjars 0.6.1 2014-08-22 [1] CRAN (R 3.6.3)
## XMAS * 0.0.0.9000 2022-03-23 [1] local
## XMAS2 2.1.8.3 2022-11-08 [2] local
## XML 3.99-0.3 2020-01-20 [1] CRAN (R 3.6.3)
## xml2 1.3.3 2021-11-30 [2] CRAN (R 3.6.3)
## xtable 1.8-4 2019-04-21 [1] CRAN (R 3.6.3)
## XVector 0.26.0 2019-10-29 [2] Bioconductor
## xviz * 1.1.0 2021-01-14 [1] local
## yaml 2.2.2 2022-01-25 [1] CRAN (R 3.6.3)
## zlibbioc 1.32.0 2019-10-29 [2] Bioconductor
## zoo 1.8-9 2021-03-09 [1] CRAN (R 3.6.3)
##
## [1] /share/home/tongbangzhuo/R/x86_64-pc-linux-gnu-library/3.6
## [2] /opt/R-3.6.3/lib/R/library
##
## R ── Package was removed from disk.
##
## ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────